

Aclacinomycin A sensitizes K562 chronic myeloid leukemia cells to imatinib through p38MAPK-mediated erythroid differentiation
- PMID: 23613979
- PMCID: PMC3629111
- DOI: 10.1371/journal.pone.0061939
Aclacinomycin A sensitizes K562 chronic myeloid leukemia cells to imatinib through p38MAPK-mediated erythroid differentiation
Erratum in
-
Correction: Aclacinomycin A Sensitizes K562 Chronic Myeloid Leukemia Cells to Imatinib through p38MAPK-Mediated Erythroid Differentiation.PLoS One. 2017 Oct 11;12(10):e0186528. doi: 10.1371/journal.pone.0186528. eCollection 2017. PLoS One. 2017. PMID: 29020096 Free PMC article.
Abstract
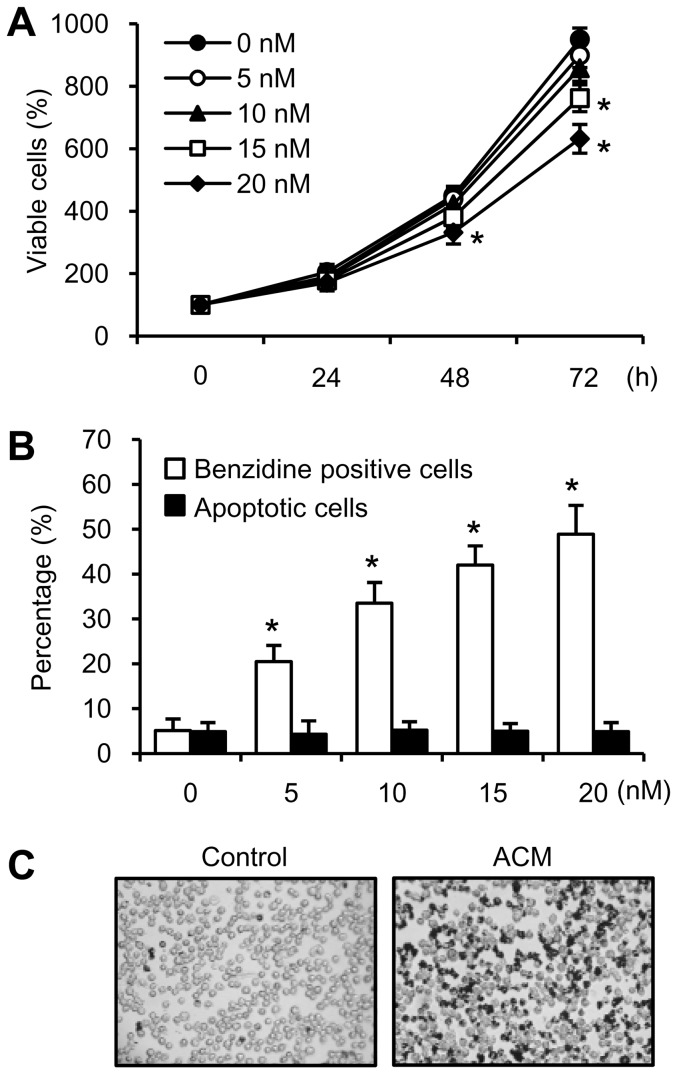
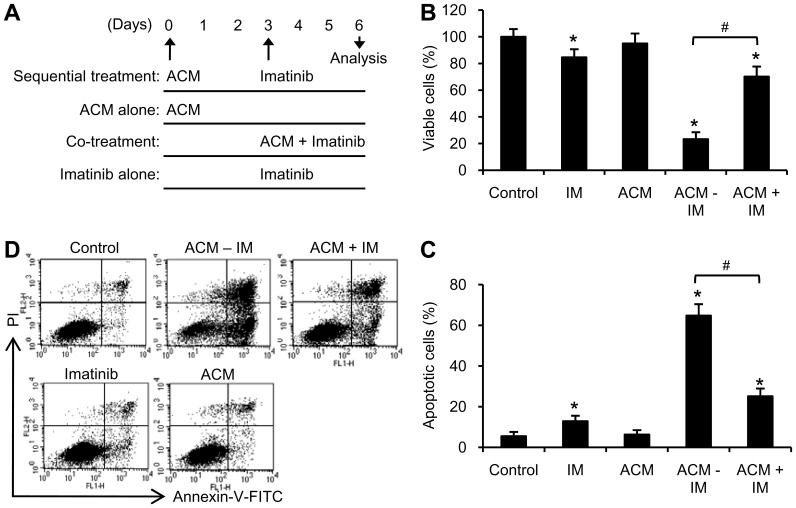
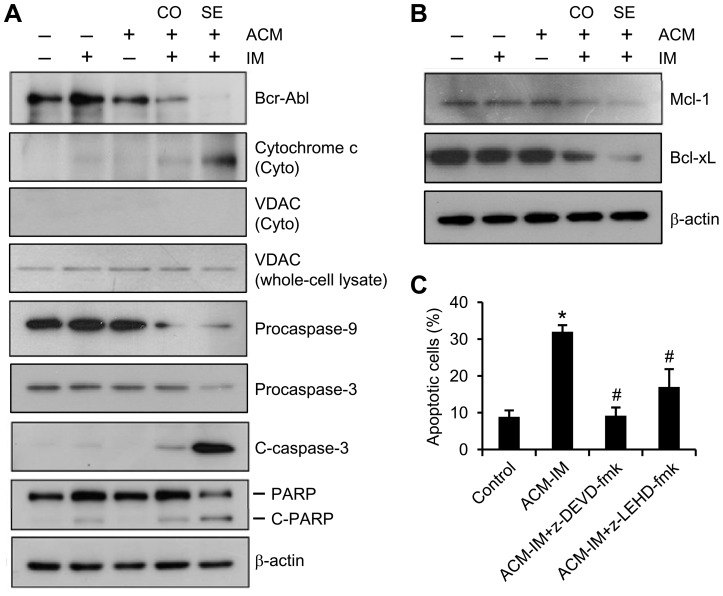
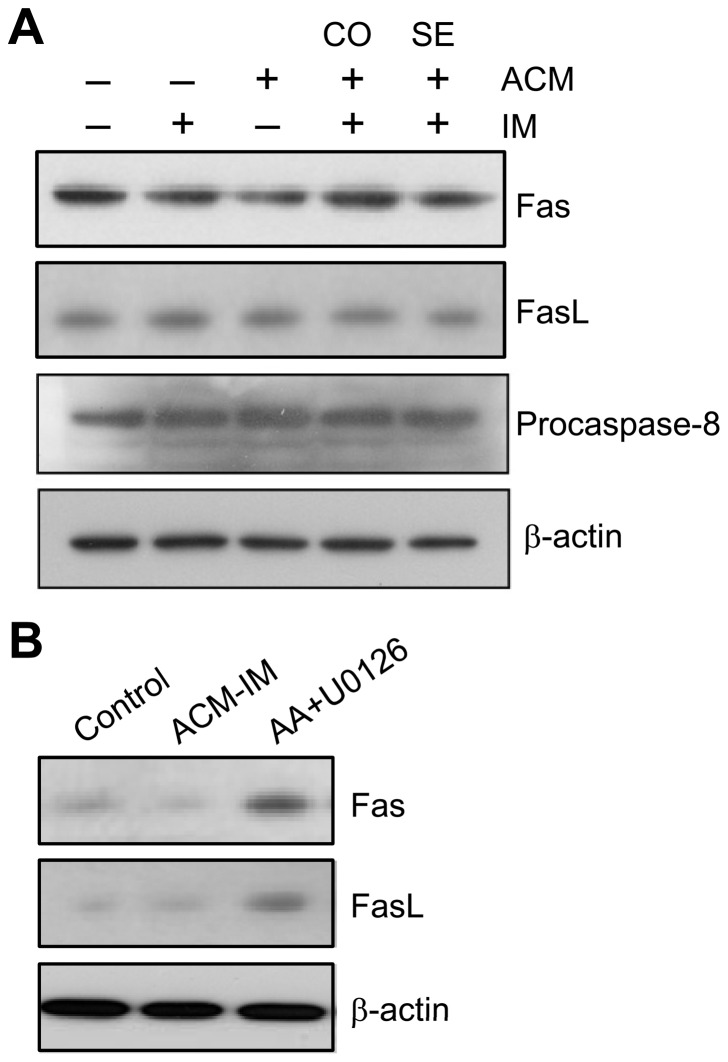
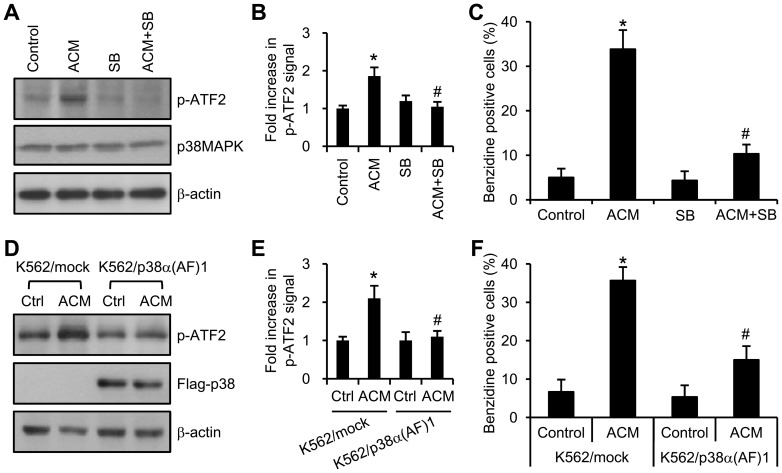
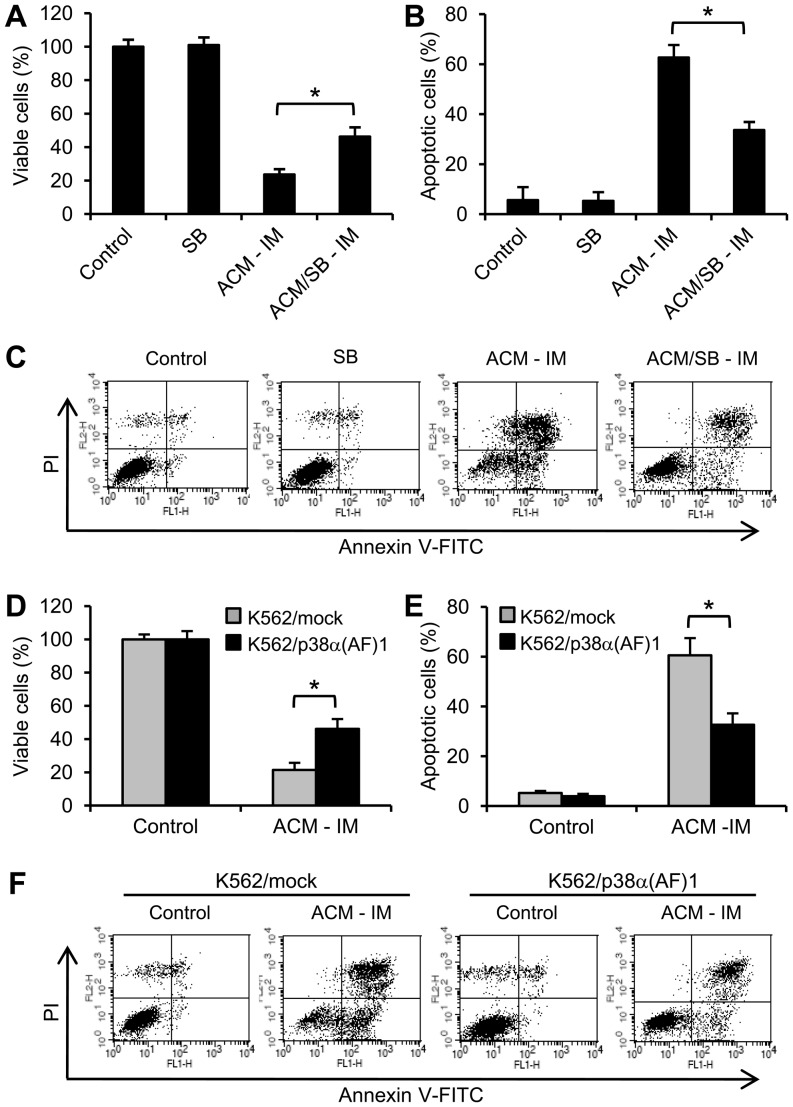
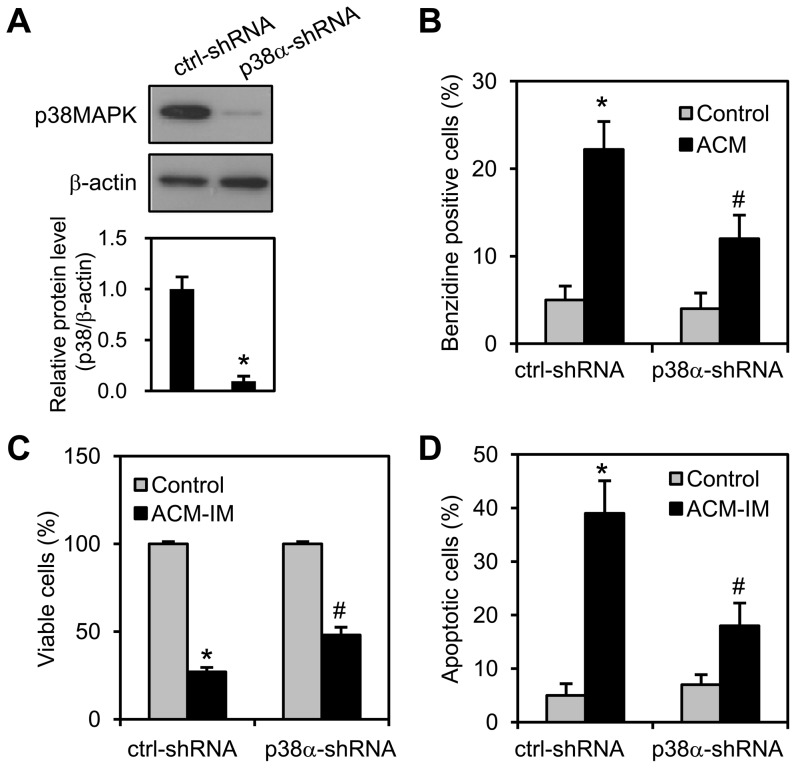
Expression of oncogenic Bcr-Abl inhibits cell differentiation of hematopoietic stem/progenitor cells in chronic myeloid leukemia (CML). Differentiation therapy is considered to be a new strategy for treating this type of leukemia. Aclacinomycin A (ACM) is an antitumor antibiotic. Previous studies have shown that ACM induced erythroid differentiation of CML cells. In this study, we investigate the effect of ACM on the sensitivity of human CML cell line K562 to Bcr-Abl specific inhibitor imatinib (STI571, Gleevec). We first determined the optimal concentration of ACM for erythroid differentiation but not growth inhibition and apoptosis in K562 cells. Then, pretreatment with this optimal concentration of ACM followed by a minimally toxic concentration of imatinib strongly induced growth inhibition and apoptosis compared to that with simultaneous co-treatment, indicating that ACM-induced erythroid differentiation sensitizes K562 cells to imatinib. Sequential treatment with ACM and imatinib induced Bcr-Abl down-regulation, cytochrome c release into the cytosol, and caspase-3 activation, as well as decreased Mcl-1 and Bcl-xL expressions, but did not affect Fas ligand/Fas death receptor and procaspase-8 expressions. ACM/imatinib sequential treatment-induced apoptosis was suppressed by a caspase-9 inhibitor and a caspase-3 inhibitor, indicating that the caspase cascade is involved in this apoptosis. Furthermore, we demonstrated that ACM induced erythroid differentiation through the p38 mitogen-activated protein kinase (MAPK) pathway. The inhibition of erythroid differentiation by p38MAPK inhibitor SB202190, p38MAPK dominant negative mutant or p38MAPK shRNA knockdown, reduced the ACM/imatinib sequential treatment-mediated growth inhibition and apoptosis. These results suggest that differentiated K562 cells induced by ACM-mediated p38MAPK pathway become more sensitive to imatinib and result in down-regulations of Bcr-Abl and anti-apoptotic proteins, growth inhibition and apoptosis. These results provided a potential management by which ACM might have a crucial impact on increasing sensitivity of CML cells to imatinib in the differentiation therapeutic approaches.
Conflict of interest statement
Figures
Similar articles
-
Apoptosis and erythroid differentiation triggered by Bcr-Abl inhibitors in CML cell lines are fully distinguishable processes that exhibit different sensitivity to caspase inhibition.Oncogene. 2007 Apr 12;26(17):2445-58. doi: 10.1038/sj.onc.1210034. Epub 2006 Oct 9. Oncogene. 2007. PMID: 17043649
-
Activin A induction of erythroid differentiation sensitizes K562 chronic myeloid leukemia cells to a subtoxic concentration of imatinib.Am J Physiol Cell Physiol. 2014 Jan 1;306(1):C37-44. doi: 10.1152/ajpcell.00130.2013. Epub 2013 Oct 2. Am J Physiol Cell Physiol. 2014. PMID: 24088895
-
Activin A downregulates the CD69-MT2A axis via p38MAPK to induce erythroid differentiation that sensitizes BCR-ABL-positive cells to imatinib.Exp Cell Res. 2022 Aug 15;417(2):113219. doi: 10.1016/j.yexcr.2022.113219. Epub 2022 May 26. Exp Cell Res. 2022. PMID: 35643179
-
FoxO tumor suppressors and BCR-ABL-induced leukemia: a matter of evasion of apoptosis.Biochim Biophys Acta. 2008 Jan;1785(1):63-84. doi: 10.1016/j.bbcan.2007.10.003. Epub 2007 Oct 16. Biochim Biophys Acta. 2008. PMID: 17980712 Free PMC article. Review.
-
Are off-target effects of imatinib the key to improving beta-cell function in diabetes?Ups J Med Sci. 2022 Sep 14;127. doi: 10.48101/ujms.v127.8841. eCollection 2022. Ups J Med Sci. 2022. PMID: 36187072 Free PMC article. Review.
Cited by
-
Gasotransmitters: Potential Therapeutic Molecules of Fibrotic Diseases.Oxid Med Cell Longev. 2021 Sep 20;2021:3206982. doi: 10.1155/2021/3206982. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34594474 Free PMC article. Review.
-
Spred2 modulates the erythroid differentiation induced by imatinib in chronic myeloid leukemia cells.PLoS One. 2015 Feb 17;10(2):e0117573. doi: 10.1371/journal.pone.0117573. eCollection 2015. PLoS One. 2015. PMID: 25688862 Free PMC article.
-
Arsenic sulfide nanoformulation induces erythroid differentiation in chronic myeloid leukemia cells through degradation of BCR-ABL.Int J Nanomedicine. 2019 Jul 22;14:5581-5594. doi: 10.2147/IJN.S207298. eCollection 2019. Int J Nanomedicine. 2019. PMID: 31413564 Free PMC article.
-
miR-150 inhibits terminal erythroid proliferation and differentiation.Oncotarget. 2015 Dec 15;6(40):43033-47. doi: 10.18632/oncotarget.5824. Oncotarget. 2015. PMID: 26543232 Free PMC article.
-
Olefin cross metathesis based de novo synthesis of a partially protected L-amicetose and a fully protected L-cinerulose derivative.Beilstein J Org Chem. 2014 May 6;10:1023-31. doi: 10.3762/bjoc.10.102. eCollection 2014. Beilstein J Org Chem. 2014. PMID: 24991253 Free PMC article.
References
-
- Deininger MW, Goldman JM, Melo JV (2000) The molecular biology of chronic myeloid leukemia. Blood 96: 3343–3356. - PubMed
-
- Holyoake DT (2001) Recent advances in the molecular and cellular biology of chronic myeloid leukaemia: lessons to be learned from the laboratory. Br J Haematol 113: 11–23. - PubMed
-
- Clarkson B, Strife A, Wisniewski D, Lambek CL, Liu C (2003) Chronic myelogenous leukemia as a paradigm of early cancer and possible curative strategies. Leukemia 17: 1211–1262. - PubMed
-
- Mauro MJ, Deininger MW (2006) Chronic myeloid leukemia in 2006: a perspective. Heamatologica 91: 152–157. - PubMed
-
- Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, et al. (2002) Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 346: 645–652. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
