

Extensive transcriptional heterogeneity revealed by isoform profiling
- PMID: 23615609
- PMCID: PMC3705217
- DOI: 10.1038/nature12121
Extensive transcriptional heterogeneity revealed by isoform profiling
Abstract
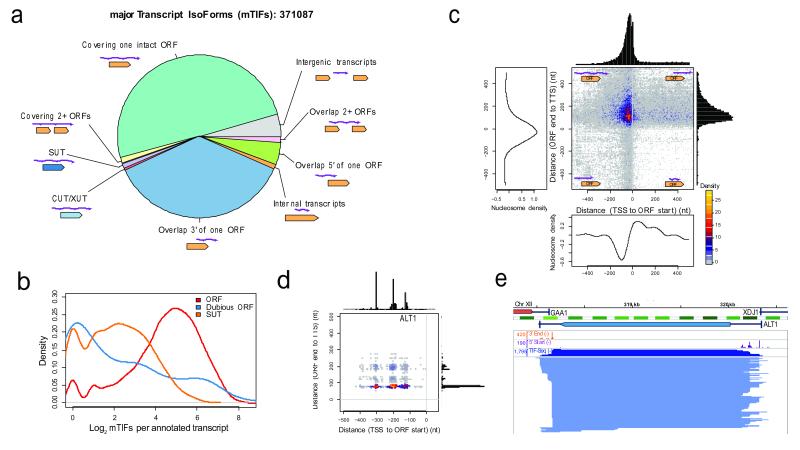
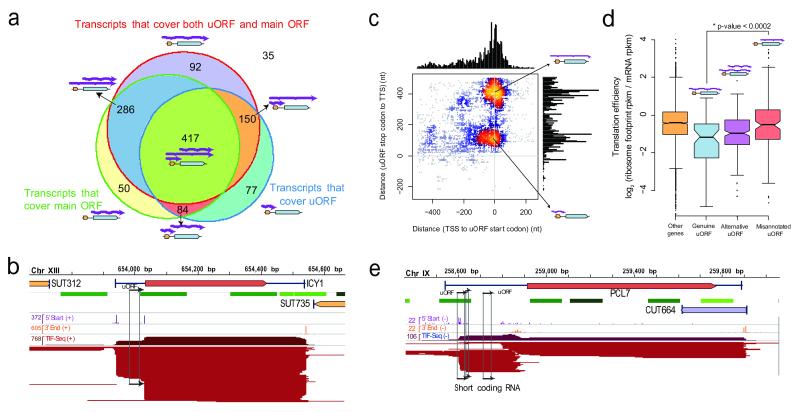
Transcript function is determined by sequence elements arranged on an individual RNA molecule. Variation in transcripts can affect messenger RNA stability, localization and translation, or produce truncated proteins that differ in localization or function. Given the existence of overlapping, variable transcript isoforms, determining the functional impact of the transcriptome requires identification of full-length transcripts, rather than just the genomic regions that are transcribed. Here, by jointly determining both transcript ends for millions of RNA molecules, we reveal an extensive layer of isoform diversity previously hidden among overlapping RNA molecules. Variation in transcript boundaries seems to be the rule rather than the exception, even within a single population of yeast cells. Over 26 major transcript isoforms per protein-coding gene were expressed in yeast. Hundreds of short coding RNAs and truncated versions of proteins are concomitantly encoded by alternative transcript isoforms, increasing protein diversity. In addition, approximately 70% of genes express alternative isoforms that vary in post-transcriptional regulatory elements, and tandem genes frequently produce overlapping or even bicistronic transcripts. This extensive transcript diversity is generated by a relatively simple eukaryotic genome with limited splicing, and within a genetically homogeneous population of cells. Our findings have implications for genome compaction, evolution and phenotypic diversity between single cells. These data also indicate that isoform diversity as well as RNA abundance should be considered when assessing the functional repertoire of genomes.
Figures


Comment in
-
Molecular biology: The ends justify the means.Nature. 2013 May 2;497(7447):48-9. doi: 10.1038/nature12098. Epub 2013 Apr 24. Nature. 2013. PMID: 23615607 No abstract available.
References
-
- Carlson M, Botstein D. Two differentially regulated mRNAs with different 5′ ends encode secreted with intracellular forms of yeast invertase. Cell. 1982;28:145–154. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
