

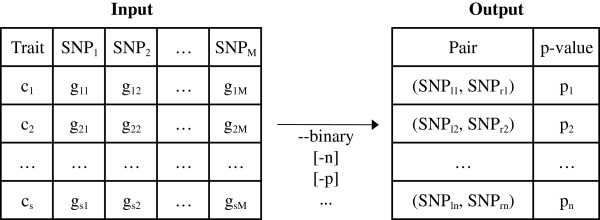
An efficient algorithm to perform multiple testing in epistasis screening
- PMID: 23617239
- PMCID: PMC3648350
- DOI: 10.1186/1471-2105-14-138
An efficient algorithm to perform multiple testing in epistasis screening
Abstract
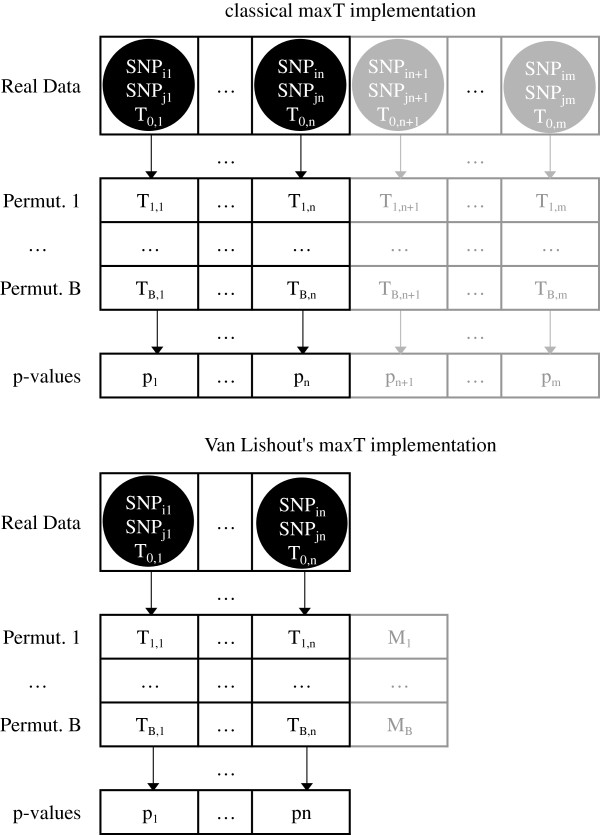
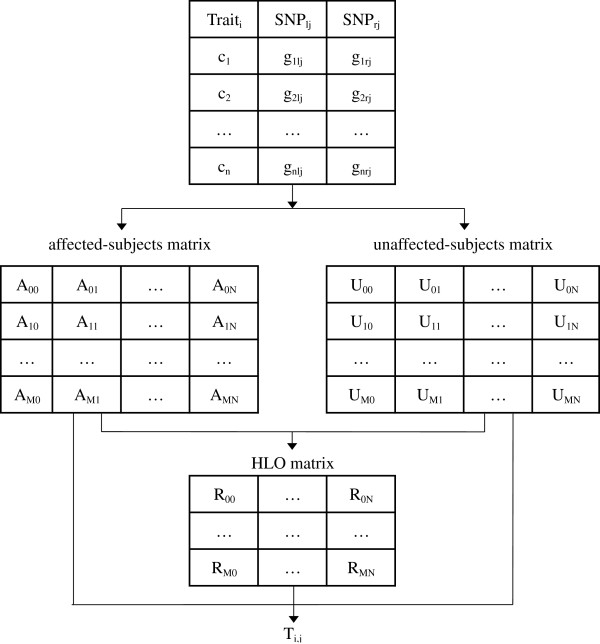
Background: Research in epistasis or gene-gene interaction detection for human complex traits has grown over the last few years. It has been marked by promising methodological developments, improved translation efforts of statistical epistasis to biological epistasis and attempts to integrate different omics information sources into the epistasis screening to enhance power. The quest for gene-gene interactions poses severe multiple-testing problems. In this context, the maxT algorithm is one technique to control the false-positive rate. However, the memory needed by this algorithm rises linearly with the amount of hypothesis tests. Gene-gene interaction studies will require a memory proportional to the squared number of SNPs. A genome-wide epistasis search would therefore require terabytes of memory. Hence, cache problems are likely to occur, increasing the computation time. In this work we present a new version of maxT, requiring an amount of memory independent from the number of genetic effects to be investigated. This algorithm was implemented in C++ in our epistasis screening software MBMDR-3.0.3. We evaluate the new implementation in terms of memory efficiency and speed using simulated data. The software is illustrated on real-life data for Crohn's disease.
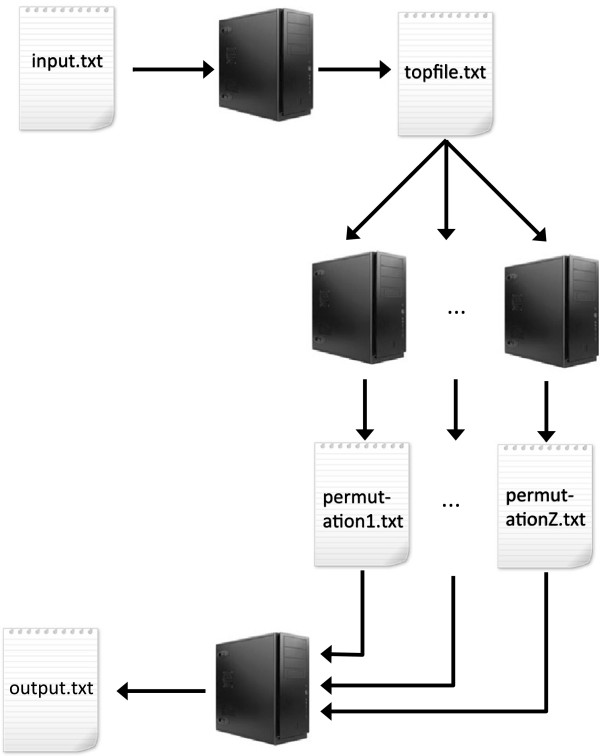
Results: In the case of a binary (affected/unaffected) trait, the parallel workflow of MBMDR-3.0.3 analyzes all gene-gene interactions with a dataset of 100,000 SNPs typed on 1000 individuals within 4 days and 9 hours, using 999 permutations of the trait to assess statistical significance, on a cluster composed of 10 blades, containing each four Quad-Core AMD Opteron(tm) Processor 2352 2.1 GHz. In the case of a continuous trait, a similar run takes 9 days. Our program found 14 SNP-SNP interactions with a multiple-testing corrected p-value of less than 0.05 on real-life Crohn's disease (CD) data.
Conclusions: Our software is the first implementation of the MB-MDR methodology able to solve large-scale SNP-SNP interactions problems within a few days, without using much memory, while adequately controlling the type I error rates. A new implementation to reach genome-wide epistasis screening is under construction. In the context of Crohn's disease, MBMDR-3.0.3 could identify epistasis involving regions that are well known in the field and could be explained from a biological point of view. This demonstrates the power of our software to find relevant phenotype-genotype higher-order associations.
Figures
References
-
- Van Steen K. Traveling the world of gene-gene interactions. Brief Bioinform. 2011;13:1–19. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
