

Vascular-targeted therapies for Duchenne muscular dystrophy
- PMID: 23618411
- PMCID: PMC3651321
- DOI: 10.1186/2044-5040-3-9
Vascular-targeted therapies for Duchenne muscular dystrophy
Abstract
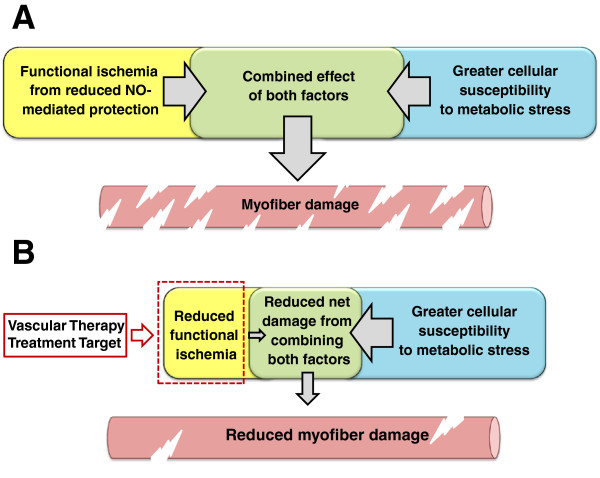
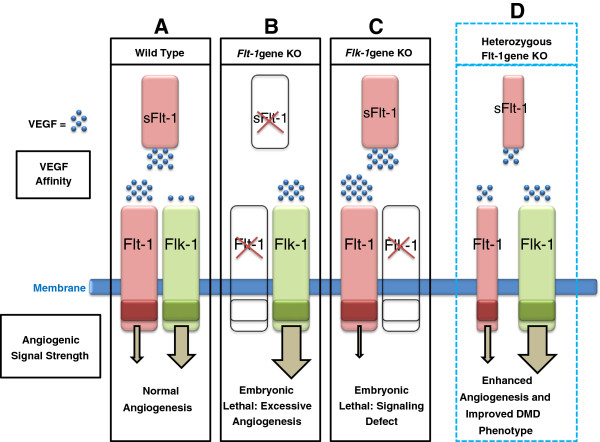
Duchenne muscular dystrophy (DMD) is the most common muscular dystrophy and an X-linked recessive, progressive muscle wasting disease caused by the absence of a functional dystrophin protein. Dystrophin has a structural role as a cytoskeletal stabilization protein and protects cells against contraction-induced damage. Dystrophin also serves a signaling role through mechanotransduction of forces and localization of neuronal nitric oxide synthase (nNOS), which produces nitric oxide (NO) to facilitate vasorelaxation. In DMD, the signaling defects produce inadequate tissue perfusion caused by functional ischemia due to a diminished ability to respond to shear stress induced endothelium-dependent dilation. Additionally, the structural defects seen in DMD render myocytes with an increased susceptibility to mechanical stress. The combination of both defects is necessary to generate myocyte damage, which induces successive rounds of myofiber degeneration and regeneration, loss of calcium homeostasis, chronic inflammatory response, fibrosis, and myonecrosis. In individuals with DMD, these processes inevitably cause loss of ambulation shortly after the first decade and an abbreviated life with death in the third or fourth decade due to cardio-respiratory anomalies. There is no known cure for DMD, and although the culpable gene has been identified for more than twenty years, research on treatments has produced few clinically relevant results. Several recent studies on novel DMD therapeutics are vascular targeted and focused on attenuating the inherent functional ischemia. One approach improves vasorelaxation capacity through pharmaceutical inhibition of either phosphodiesterase 5 (PDE5) or angiotensin-converting enzyme (ACE). Another approach increases the density of the underlying vascular network by inducing angiogenesis, and this has been accomplished through either direct delivery of vascular endothelial growth factor (VEGF) or by downregulating the VEGF decoy-receptor type 1 (VEGFR-1 or Flt-1). The pro-angiogenic approaches also seem to be pro-myogenic and could resolve the age-related decline in satellite cell (SC) quantity seen in mdx models through expansion of the SC juxtavascular niche. Here we review these four vascular targeted treatment strategies for DMD and discuss mechanisms, proof of concept, and the potential for clinical relevance associated with each therapy.
Figures
Similar articles
-
Angiogenesis as a novel therapeutic strategy for Duchenne muscular dystrophy through decreased ischemia and increased satellite cells.Front Physiol. 2014 Feb 18;5:50. doi: 10.3389/fphys.2014.00050. eCollection 2014. Front Physiol. 2014. PMID: 24600399 Free PMC article. Review.
-
Vascular therapy for Duchenne muscular dystrophy (DMD).Fac Rev. 2023 Feb 21;12:3. doi: 10.12703/r/12-3. eCollection 2023. Fac Rev. 2023. PMID: 36873982 Free PMC article. Review.
-
Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy.J Pathol. 2012 Sep;228(1):77-87. doi: 10.1002/path.4054. Epub 2012 Jul 18. J Pathol. 2012. PMID: 22653783 Free PMC article.
-
Nitric oxide-dependent attenuation of noradrenaline-induced vasoconstriction is impaired in the canine model of Duchenne muscular dystrophy.J Physiol. 2018 Nov;596(21):5199-5216. doi: 10.1113/JP275672. Epub 2018 Sep 20. J Physiol. 2018. PMID: 30152022 Free PMC article.
-
Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy.Am J Cardiovasc Drugs. 2011 Oct 1;11(5):287-94. doi: 10.2165/11594070-000000000-00000. Am J Cardiovasc Drugs. 2011. PMID: 21812510 Review.
Cited by
-
Age-Dependent Dysregulation of Muscle Vasculature and Blood Flow Recovery after Hindlimb Ischemia in the mdx Model of Duchenne Muscular Dystrophy.Biomedicines. 2021 Apr 27;9(5):481. doi: 10.3390/biomedicines9050481. Biomedicines. 2021. PMID: 33925757 Free PMC article.
-
Targeting the Muscle-Bone Unit: Filling Two Needs with One Deed in the Treatment of Duchenne Muscular Dystrophy.Curr Osteoporos Rep. 2018 Oct;16(5):541-553. doi: 10.1007/s11914-018-0468-2. Curr Osteoporos Rep. 2018. PMID: 30225627 Review.
-
HIF-hypoxia signaling in skeletal muscle physiology and fibrosis.J Cell Commun Signal. 2020 Jun;14(2):147-158. doi: 10.1007/s12079-020-00553-8. Epub 2020 Feb 22. J Cell Commun Signal. 2020. PMID: 32088838 Free PMC article. Review.
-
Characterization of the Ang/Tie2 Signaling Pathway in the Diaphragm Muscle of DMD Mice.Biomedicines. 2023 Aug 14;11(8):2265. doi: 10.3390/biomedicines11082265. Biomedicines. 2023. PMID: 37626761 Free PMC article.
-
Cardiovascular phenotype of the Dmdmdx rat - a suitable animal model for Duchenne muscular dystrophy.Dis Model Mech. 2021 Feb 22;14(2):dmm047704. doi: 10.1242/dmm.047704. Dis Model Mech. 2021. PMID: 33619211 Free PMC article.
References
-
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
