

Arachidonic acid-induced dilation in human coronary arterioles: convergence of signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry
- PMID: 23619744
- PMCID: PMC3698766
- DOI: 10.1161/JAHA.113.000080
Arachidonic acid-induced dilation in human coronary arterioles: convergence of signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry
Abstract
Background: Arachidonic acid (AA) and/or its enzymatic metabolites are important lipid mediators contributing to endothelium-derived hyperpolarizing factor (EDHF)-mediated dilation in multiple vascular beds, including human coronary arterioles (HCAs). However, the mechanisms of action of these lipid mediators in endothelial cells (ECs) remain incompletely defined. In this study, we investigated the role of the transient receptor potential vanilloid 4 (TRPV4) channel in AA-induced endothelial Ca(2+) response and dilation of HCAs.
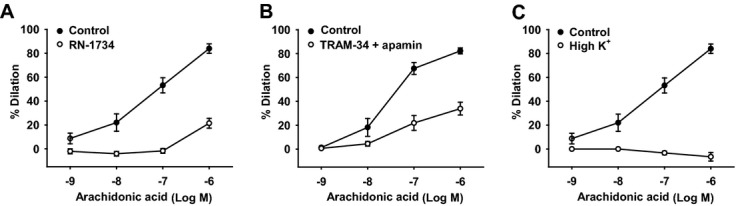
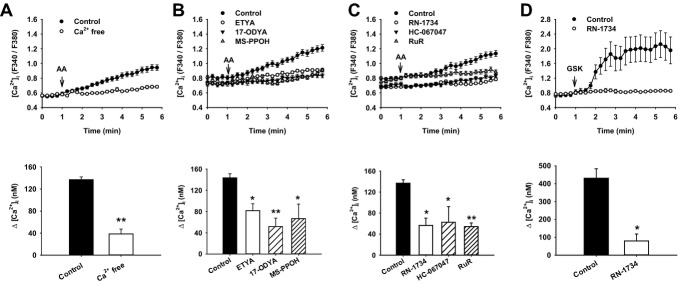
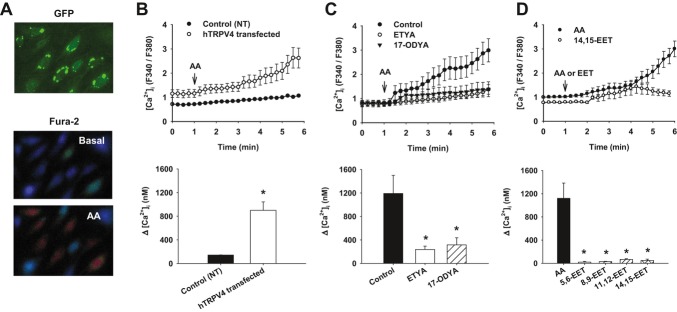
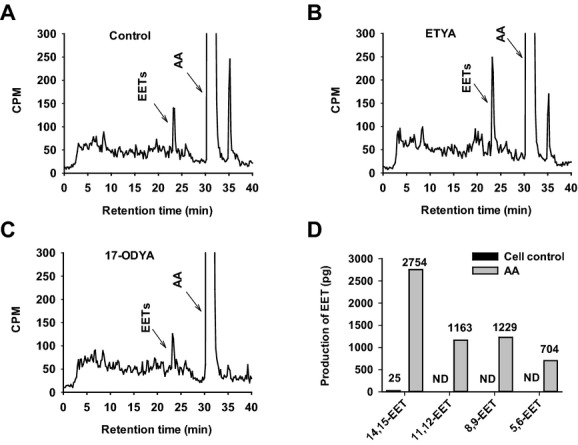
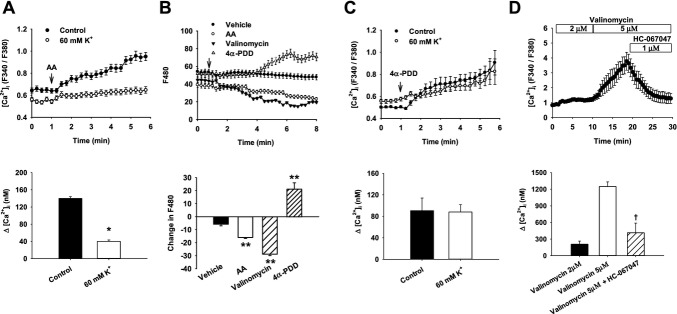
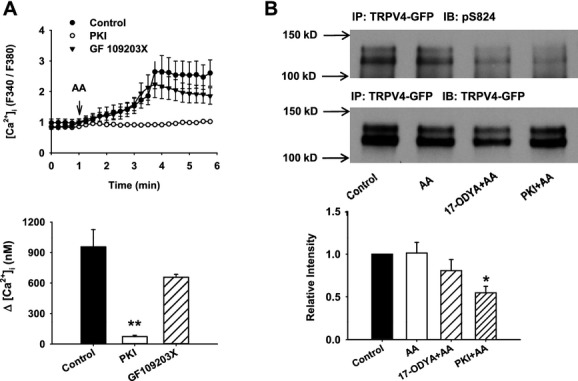
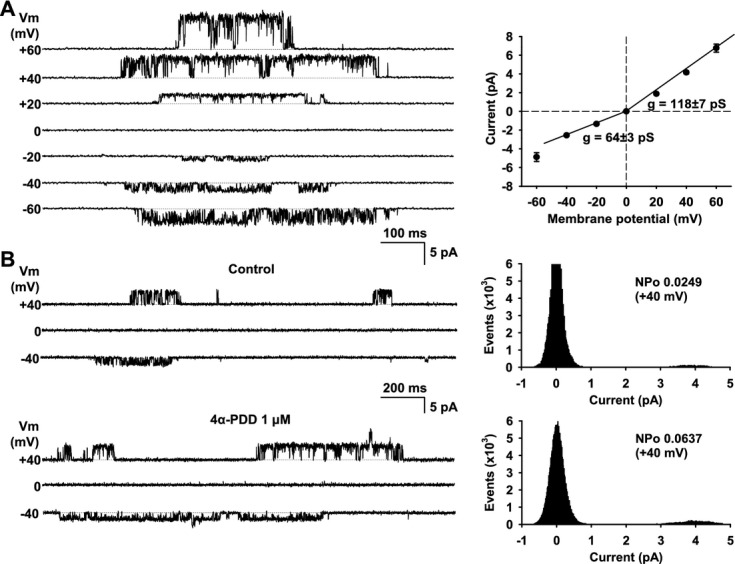
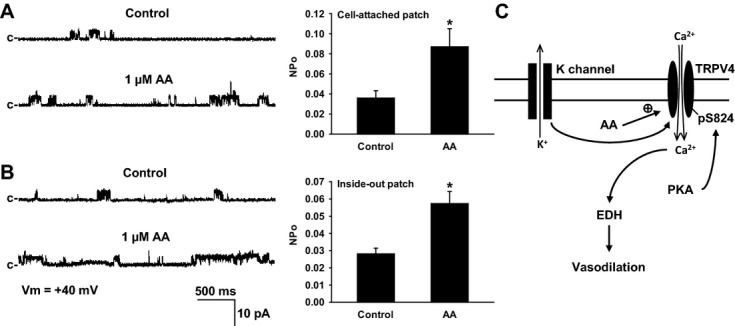
Methods and results: AA induced concentration-dependent dilation in isolated HCAs. The dilation was largely abolished by the TRPV4 antagonist RN-1734 and by inhibition of endothelial Ca(2+)-activated K(+) channels. In native and TRPV4-overexpressing human coronary artery ECs (HCAECs), AA increased intracellular Ca(2+) concentration ([Ca(2+)]i), which was mediated by TRPV4-dependent Ca(2+) entry. The AA-induced [Ca(2+)]i increase was inhibited by cytochrome P450 (CYP) inhibitors. Surprisingly, the CYP metabolites of AA, epoxyeicosatrienoic acids (EETs), were much less potent activators of TRPV4, and CYP inhibitors did not affect EET production in HCAECs. Apart from its effect on [Ca(2+)]i, AA induced endothelial hyperpolarization, and this effect was required for Ca(2+) entry through TRPV4. AA-induced and TRPV4-mediated Ca(2+) entry was also inhibited by the protein kinase A inhibitor PKI. TRPV4 exhibited a basal level of phosphorylation, which was inhibited by PKI. Patch-clamp studies indicated that AA activated TRPV4 single-channel currents in cell-attached and inside-out patches of HCAECs.
Conclusions: AA dilates HCAs through a novel mechanism involving endothelial TRPV4 channel-dependent Ca(2+) entry that requires endothelial hyperpolarization, PKA-mediated basal phosphorylation of TRPV4, and direct activation of TRPV4 channels by AA.
Figures
Similar articles
-
Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation.J Biol Chem. 2018 Apr 6;293(14):5307-5322. doi: 10.1074/jbc.M117.811075. Epub 2018 Feb 8. J Biol Chem. 2018. PMID: 29462784 Free PMC article.
-
Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: role of Ca2+ entry and mitochondrial ROS signaling.Am J Physiol Heart Circ Physiol. 2012 Feb 1;302(3):H634-42. doi: 10.1152/ajpheart.00717.2011. Epub 2011 Dec 2. Am J Physiol Heart Circ Physiol. 2012. PMID: 22140047 Free PMC article.
-
H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation.Circ Res. 2012 Feb 3;110(3):471-80. doi: 10.1161/CIRCRESAHA.111.258871. Epub 2011 Dec 8. Circ Res. 2012. PMID: 22158710 Free PMC article.
-
Endothelium-dependent cerebral artery dilation mediated by transient receptor potential and Ca2+-activated K+ channels.J Cardiovasc Pharmacol. 2011 Feb;57(2):148-53. doi: 10.1097/FJC.0b013e3181f580d9. J Cardiovasc Pharmacol. 2011. PMID: 20729757 Review.
-
Role of cytochrome P450-dependent arachidonic acid metabolites in liver physiology and pathophysiology.Prostaglandins Other Lipid Mediat. 2003 Oct;72(1-2):51-71. doi: 10.1016/s1098-8823(03)00077-7. Prostaglandins Other Lipid Mediat. 2003. PMID: 14626496 Review.
Cited by
-
Transient receptor potential vanilloid 4 (TRPV4) activation by arachidonic acid requires protein kinase A-mediated phosphorylation.J Biol Chem. 2018 Apr 6;293(14):5307-5322. doi: 10.1074/jbc.M117.811075. Epub 2018 Feb 8. J Biol Chem. 2018. PMID: 29462784 Free PMC article.
-
The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen.Int J Mol Sci. 2018 Mar 21;19(4):938. doi: 10.3390/ijms19040938. Int J Mol Sci. 2018. PMID: 29561829 Free PMC article. Review.
-
TRPV4 channels contribute to renal myogenic autoregulation in neonatal pigs.Am J Physiol Renal Physiol. 2017 Nov 1;313(5):F1136-F1148. doi: 10.1152/ajprenal.00300.2017. Epub 2017 Aug 2. Am J Physiol Renal Physiol. 2017. PMID: 28768667 Free PMC article.
-
Effects of SGLT2 inhibitors on ion channels in heart failure: focus on the endothelium.Basic Res Cardiol. 2025 Aug;120(4):779-798. doi: 10.1007/s00395-025-01115-y. Epub 2025 May 14. Basic Res Cardiol. 2025. PMID: 40366385 Free PMC article. Review.
-
Phosphorylation of distal C-terminal residues promotes TRPV4 channel activation in response to arachidonic acid.J Biol Chem. 2025 Mar;301(3):108260. doi: 10.1016/j.jbc.2025.108260. Epub 2025 Feb 3. J Biol Chem. 2025. PMID: 39909371 Free PMC article.
References
-
- Edwards G, Feletou M, Weston AH. Endothelium‐derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch. 2010; 459:863-879 - PubMed
-
- Shimokawa H. Hydrogen peroxide as an endothelium‐derived hyperpolarizing factor. Pflugers Arch. 2010; 459:915-922 - PubMed
-
- Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow‐induced dilation of human coronary arterioles. Circ Res. 2003; 92:e31-e40 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
