

Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding
- PMID: 23622248
- PMCID: PMC3650559
- DOI: 10.1016/j.cell.2013.03.043
Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding
Abstract
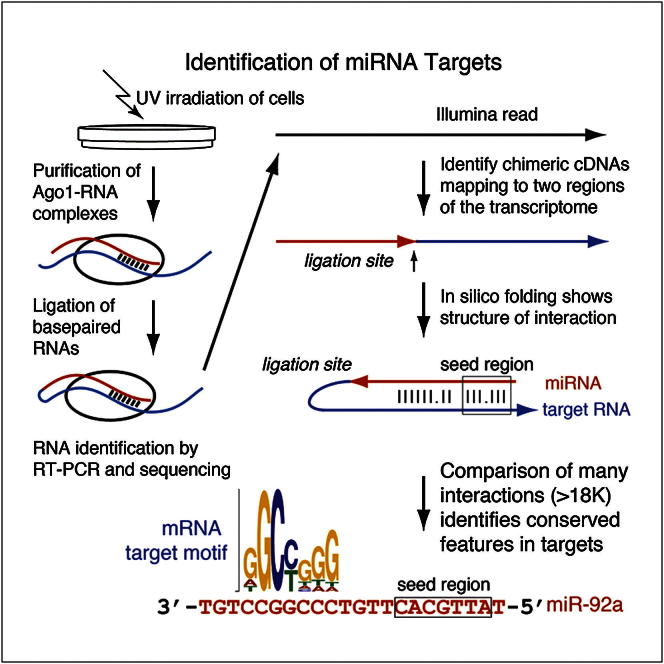
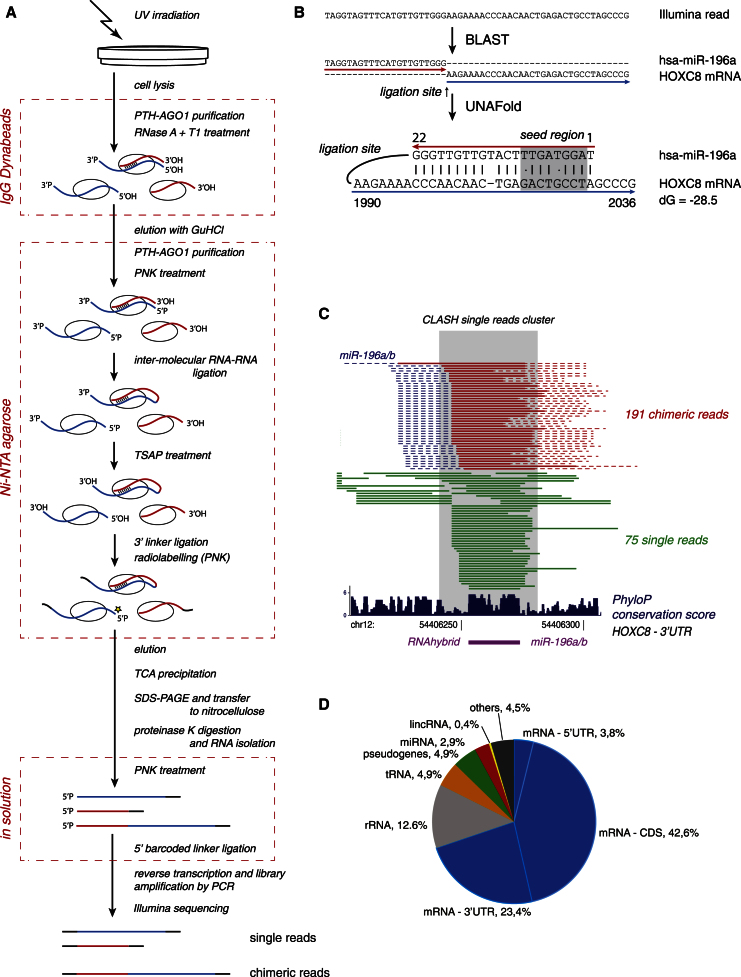
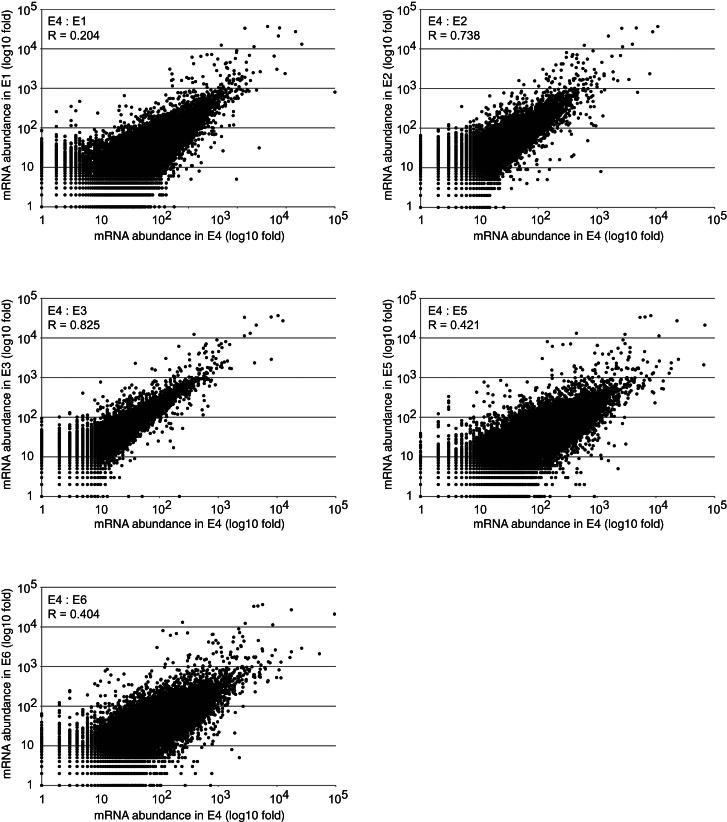
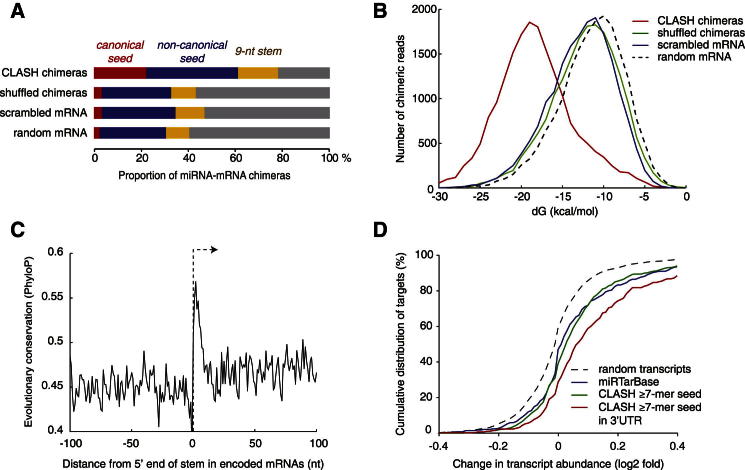
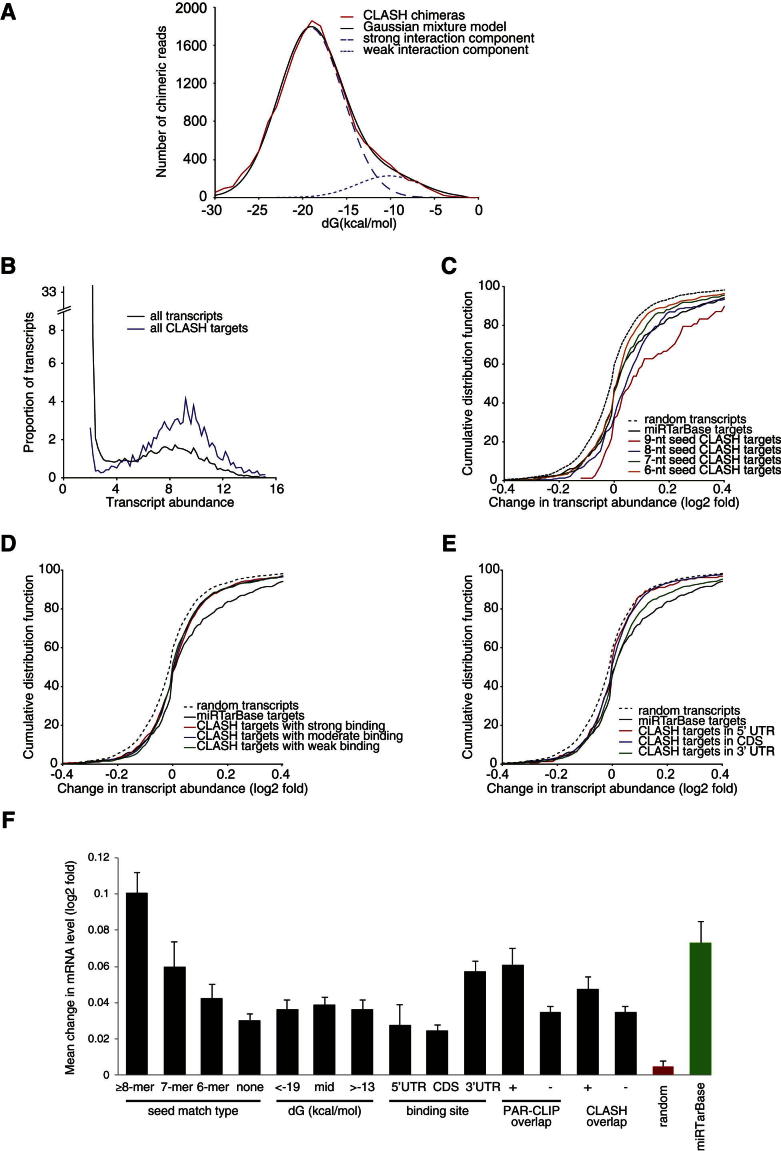
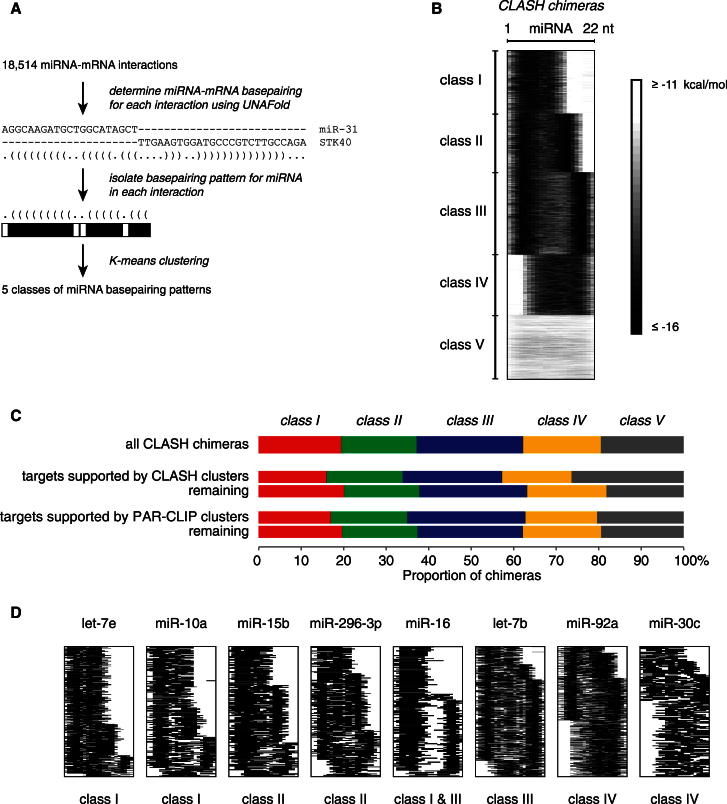
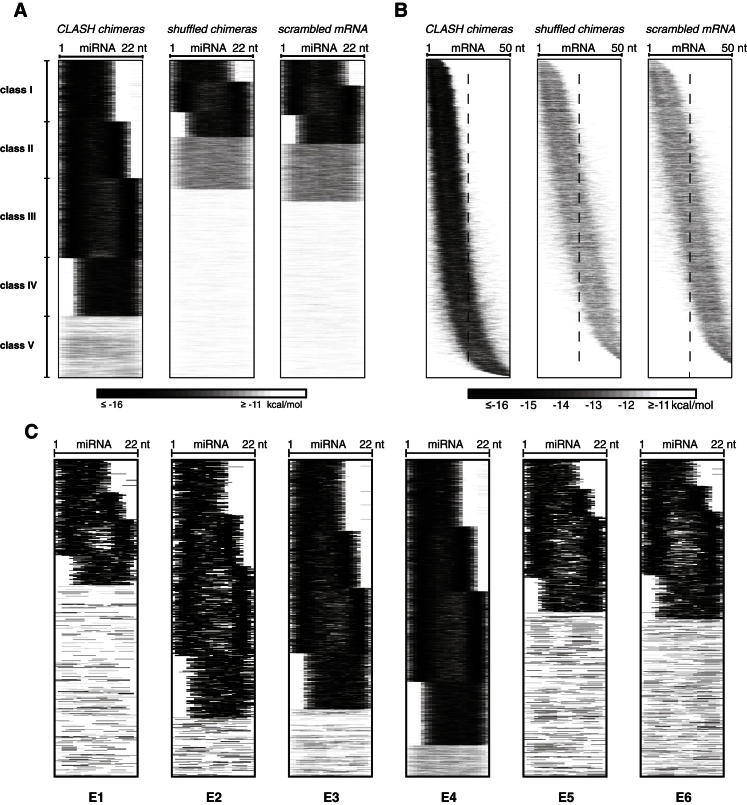
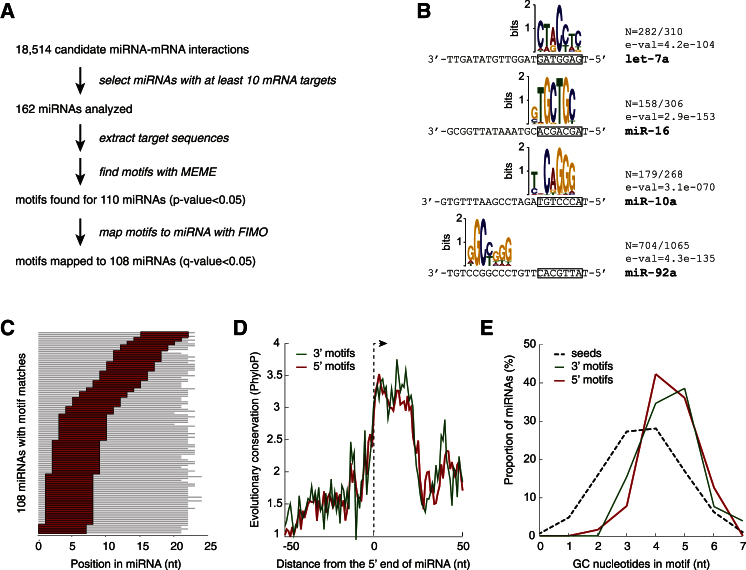
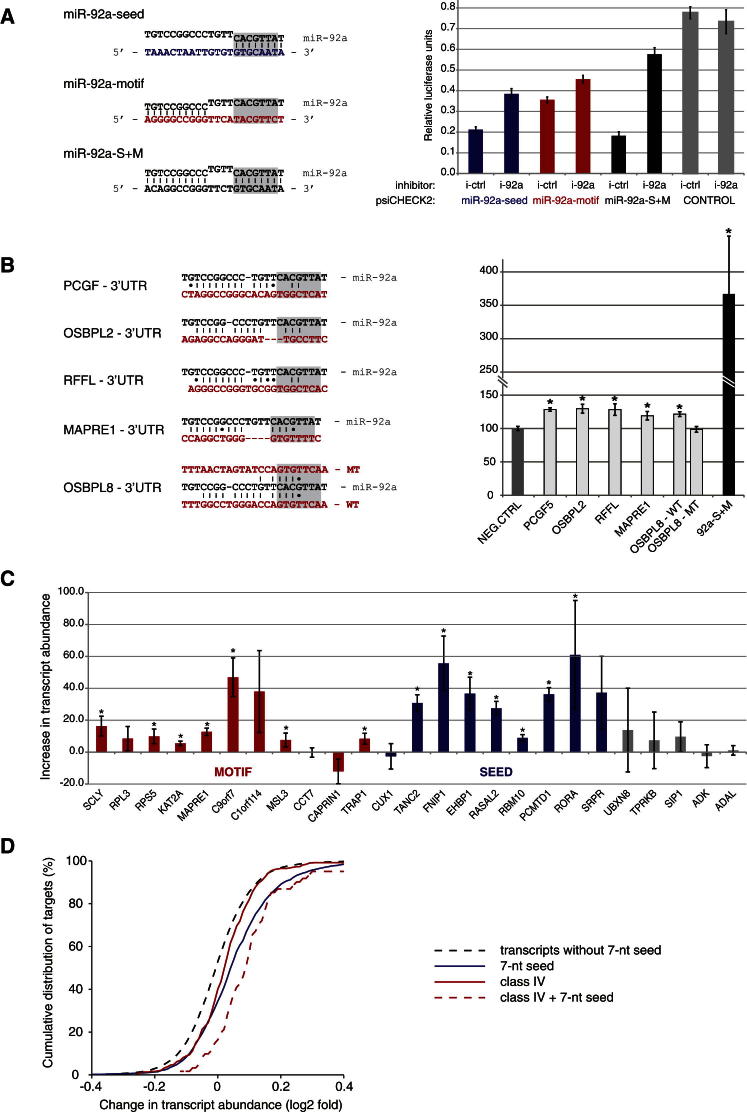
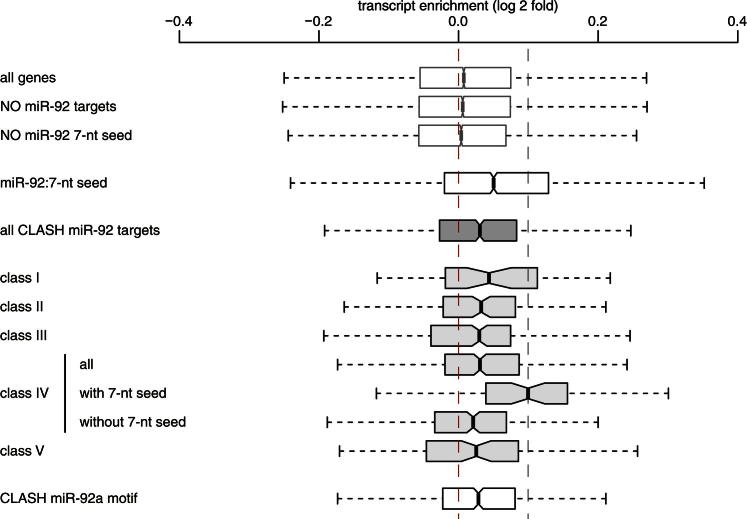
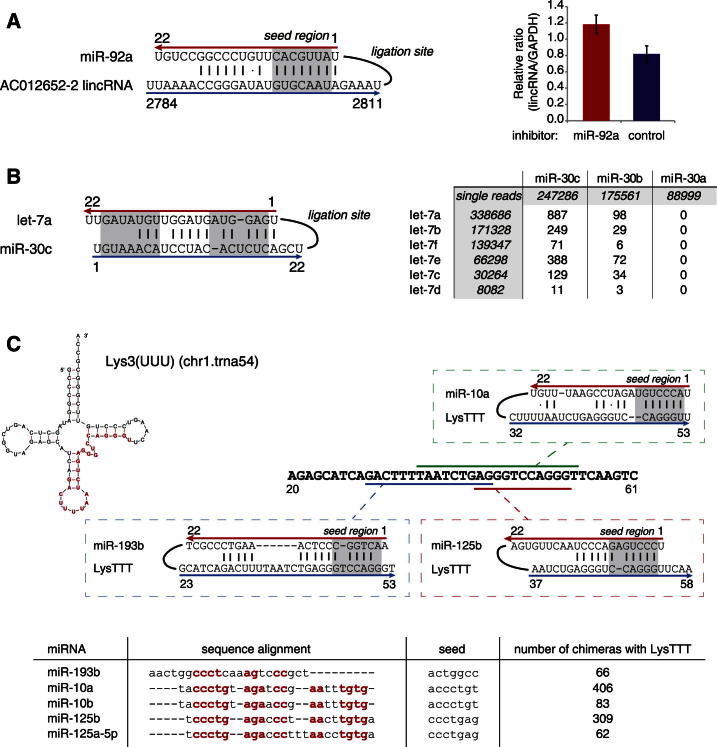
MicroRNAs (miRNAs) play key roles in gene regulation, but reliable bioinformatic or experimental identification of their targets remains difficult. To provide an unbiased view of human miRNA targets, we developed a technique for ligation and sequencing of miRNA-target RNA duplexes associated with human AGO1. Here, we report data sets of more than 18,000 high-confidence miRNA-mRNA interactions. The binding of most miRNAs includes the 5' seed region, but around 60% of seed interactions are noncanonical, containing bulged or mismatched nucleotides. Moreover, seed interactions are generally accompanied by specific, nonseed base pairing. 18% of miRNA-mRNA interactions involve the miRNA 3' end, with little evidence for 5' contacts, and some of these were functionally validated. Analyses of miRNA:mRNA base pairing showed that miRNA species systematically differ in their target RNA interactions, and strongly overrepresented motifs were found in the interaction sites of several miRNAs. We speculate that these affect the response of RISC to miRNA-target binding.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures

References
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Bailey, T.L., and Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, 28–36. - PubMed
Supplemental References
-
- Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. - PubMed
-
- Oeffinger M., Wei K.E., Rogers R., DeGrasse J.A., Chait B.T., Aitchison J.D., Rout M.P. Comprehensive analysis of diverse ribonucleoprotein complexes. Nat. Methods. 2007;4:951–956. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
