

Yunis-Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase
- PMID: 23623387
- PMCID: PMC3644641
- DOI: 10.1016/j.ajhg.2013.03.020
Yunis-Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase
Abstract
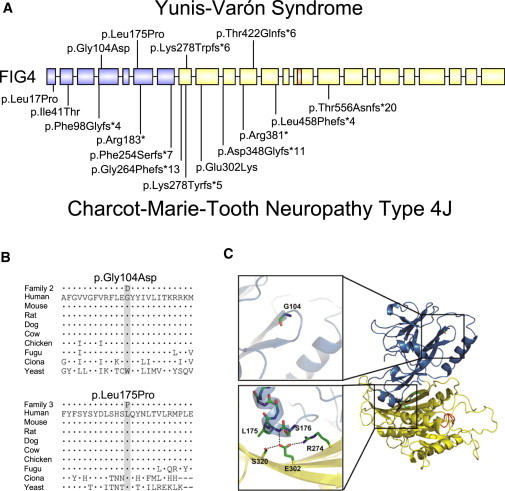
Yunis-Varón syndrome (YVS) is an autosomal-recessive disorder with cleidocranial dysplasia, digital anomalies, and severe neurological involvement. Enlarged vacuoles are found in neurons, muscle, and cartilage. By whole-exome sequencing, we identified frameshift and missense mutations of FIG4 in affected individuals from three unrelated families. FIG4 encodes a phosphoinositide phosphatase required for regulation of PI(3,5)P(2) levels, and thus endosomal trafficking and autophagy. In a functional assay, both missense substitutions failed to correct the vacuolar phenotype of Fig4-null mouse fibroblasts. Homozygous Fig4-null mice exhibit features of YVS, including neurodegeneration and enlarged vacuoles in neurons. We demonstrate that Fig4-null mice also have small skeletons with reduced trabecular bone volume and cortical thickness and that cultured osteoblasts accumulate large vacuoles. Our findings demonstrate that homozygosity or compound heterozygosity for null mutations of FIG4 is responsible for YVS, the most severe known human phenotype caused by defective phosphoinositide metabolism. In contrast, in Charcot-Marie-Tooth disease type 4J (also caused by FIG4 mutations), one of the FIG4 alleles is hypomorphic and disease is limited to the peripheral nervous system. This genotype-phenotype correlation demonstrates that absence of FIG4 activity leads to central nervous system dysfunction and extensive skeletal anomalies. Our results describe a role for PI(3,5)P(2) signaling in skeletal development and maintenance.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures





References
-
- Yunis E., Varón H. Cleidocranial dysostosis, severe micrognathism, bilateral absence of thumbs and first metatarsal bone, and distal aphalangia: a new genetic syndrome. Am. J. Dis. Child. 1980;134:649–653. - PubMed
-
- Hughes H.E., Partington M.W. Brief clinical report: the syndrome of Yunis and Varón—report of a further case. Am. J. Med. Genet. 1983;14:539–544. - PubMed
-
- Pfeiffer R.A., Diekmann L., Stock H.J. Aplasia of the thumbs and great toes as the outstanding feature of Yunis and Varon syndrome. A new entity. A new observation. Ann. Genet. 1988;31:241–243. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
