

Glutathione reductase is essential for host defense against bacterial infection
- PMID: 23623936
- PMCID: PMC3749296
- DOI: 10.1016/j.freeradbiomed.2013.04.015
Glutathione reductase is essential for host defense against bacterial infection
Abstract
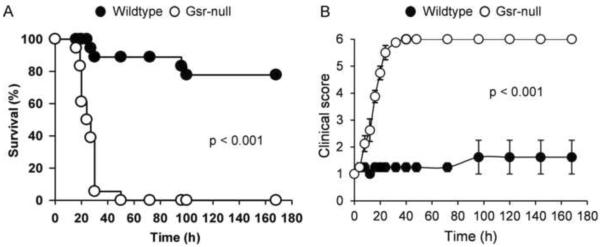
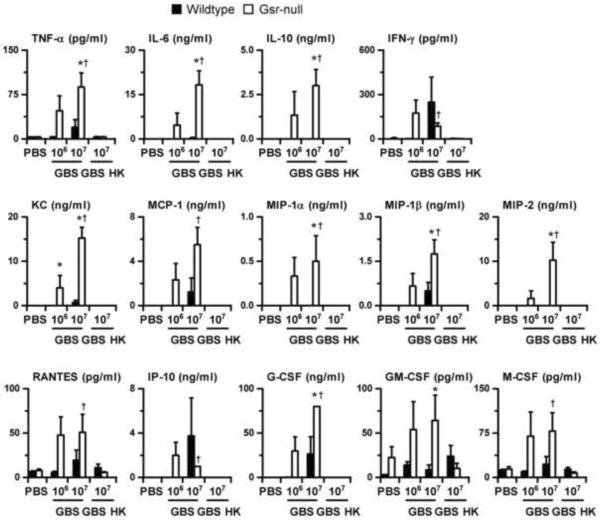
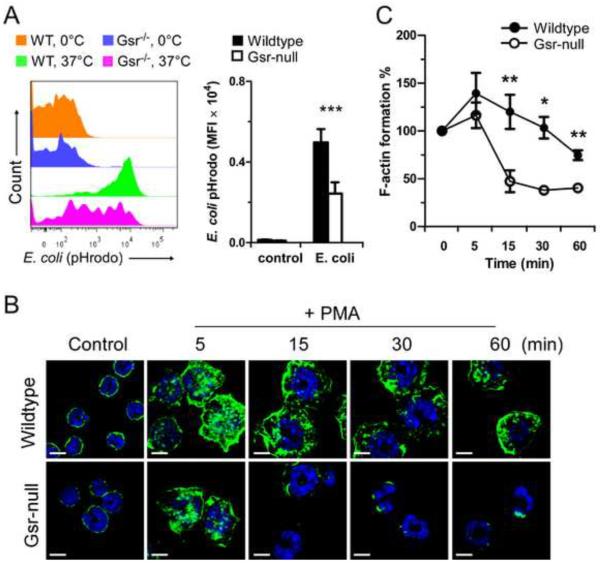
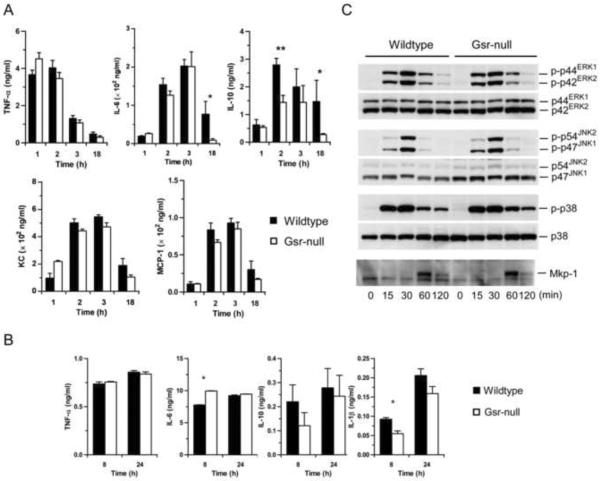
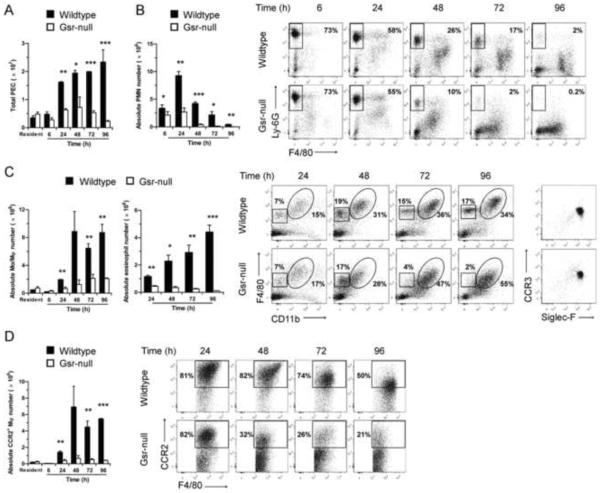
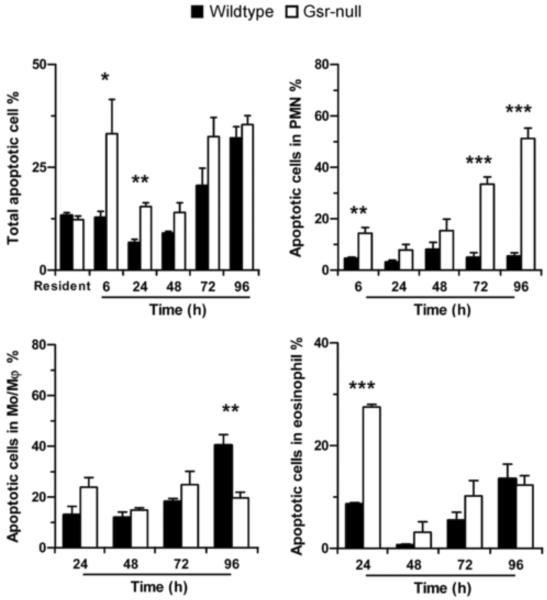
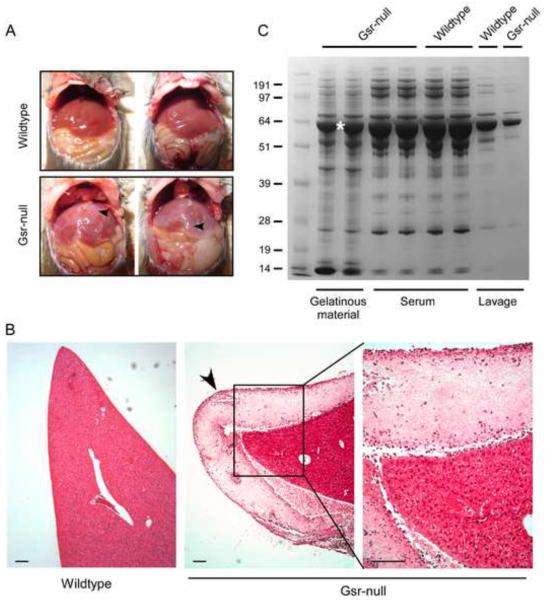
Glutathione reductase (Gsr) catalyzes the reduction of glutathione disulfide to glutathione, a major cellular antioxidant. We have recently shown that Gsr is essential for host defense against the gram-negative bacteria Escherichia coli in a mouse model of sepsis. Although we have demonstrated that Gsr is required for sustaining the oxidative burst and the development of neutrophil extracellular traps, the role of Gsr in other phagocytic functions remains unclear. It is also unclear whether Gsr-deficient mice exhibit host defense defects against gram-positive bacteria. In this study, we characterized the effects of Gsr deficiency on the innate immune responses to a gram-positive bacterium, group B Streptococcus, and to the gram-negative bacterial cell wall component lipopolysaccharide (LPS). We found that, like E. coli, group B Streptococcus resulted in a substantially more robust cytokine response and a markedly higher morbidity and mortality in Gsr-deficient mice than in wild-type mice. The increased morbidity and mortality were associated with greater bacterial burden in the Gsr-deficient mice. Interestingly, Gsr-deficient mice did not exhibit a greater sensitivity to LPS than did wild-type mice. Analysis of the neutrophils of Gsr-deficient mice revealed impaired phagocytosis. In response to thioglycollate stimulation, Gsr-deficient mice mobilized far fewer phagocytes, including neutrophils, macrophages, and eosinophils, into their peritoneal cavities than did wild-type mice. The defective phagocyte mobilization is associated with profound oxidation and aggregation of ascitic proteins, particularly albumin. Our results indicate that the oxidative defense mechanism mediated by Gsr is required for an effective innate immune response against bacteria, probably by preventing phagocyte dysfunction due to oxidative damage.
Keywords: Animal model; Bacterial infection; Free radicals; Group B Streptococcus; Host defense; Oxidative burst; Phagocytes; Redox regulation.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Reed PW. Glutathione and the hexose monophosphate shunt in phagocytizing and hydrogen peroxide-treated rat leukocytes. J. Biol. Chem. 1969;244:2459–2464. - PubMed
-
- Strauss RR, Paul BB, Jacobs AA, Sbarra AJ. The role of the phagocyte in host-parasite interactions. XIX. Leukocytic glutathione reductase and its involvement in phagocytosis. Arch. Biochem. Biophys. 1969;135:265–271. - PubMed
-
- Loos H, Roos D, Weening R, Houwerzijl J. Familial deficiency of glutathione reductase in human blood cells. Blood. 1976;48:53–62. - PubMed
-
- Roos D, Weening RS, Voetman AA, van Schaik ML, Bot AA, Meerhof LJ, Loos JA. Protection of phagocytic leukocytes by endogenous glutathione: studies in a family with glutathione reductase deficiency. Blood. 1979;53:851–866. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
