

Molecular mechanism of 17-allylamino-17-demethoxygeldanamycin (17-AAG)-induced AXL receptor tyrosine kinase degradation
- PMID: 23629654
- PMCID: PMC3682548
- DOI: 10.1074/jbc.M112.439422
Molecular mechanism of 17-allylamino-17-demethoxygeldanamycin (17-AAG)-induced AXL receptor tyrosine kinase degradation
Abstract
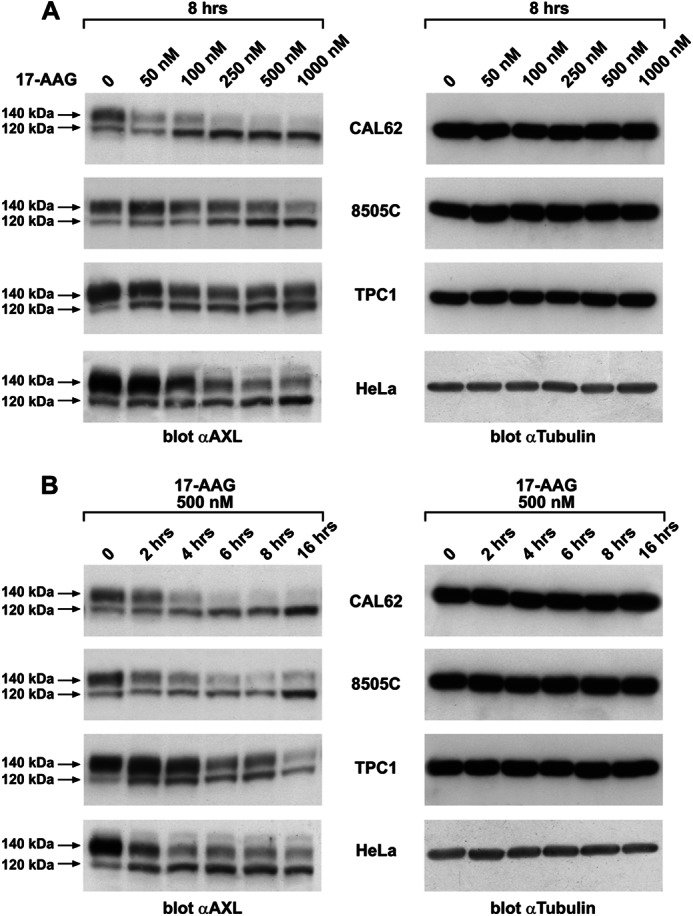
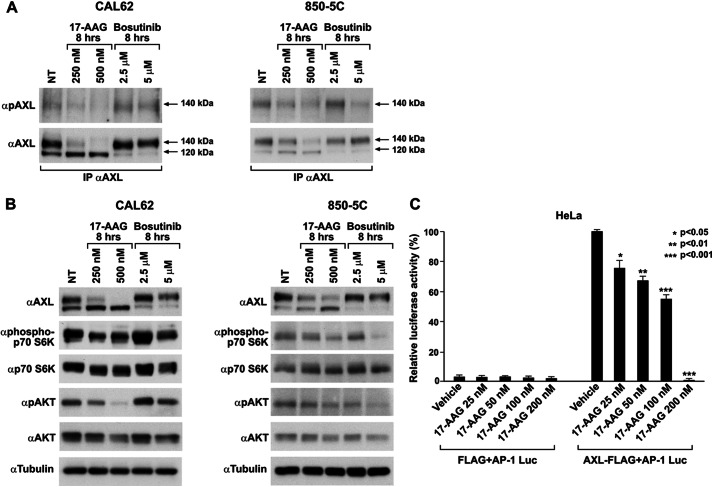
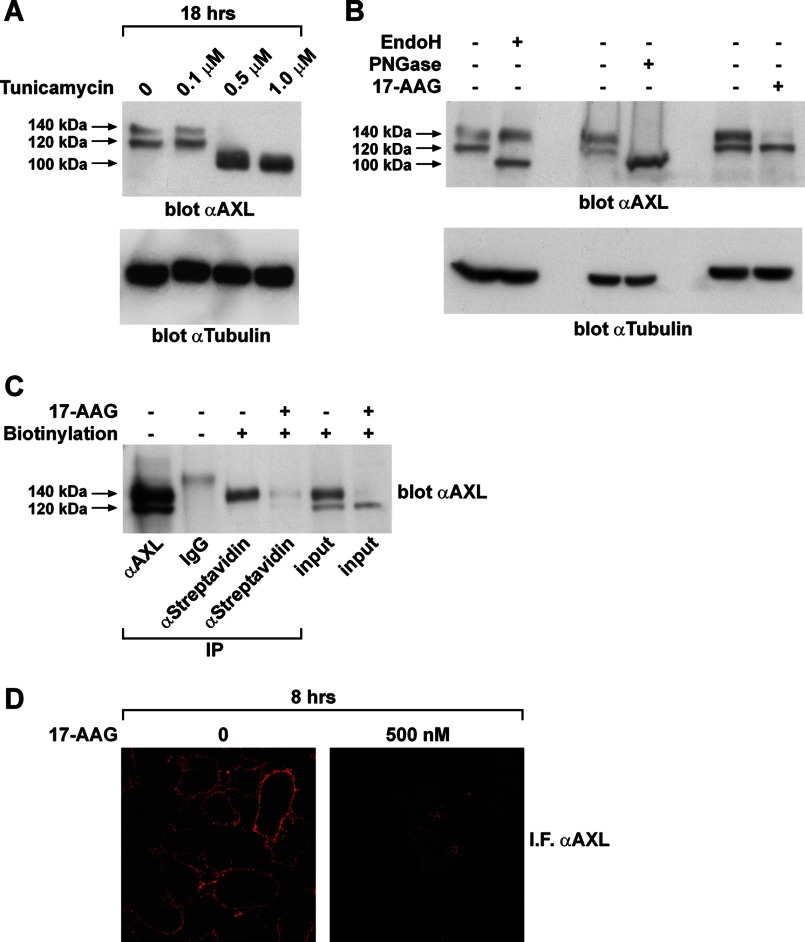
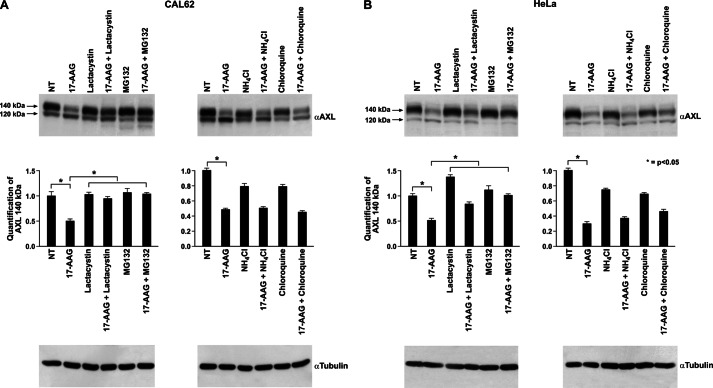
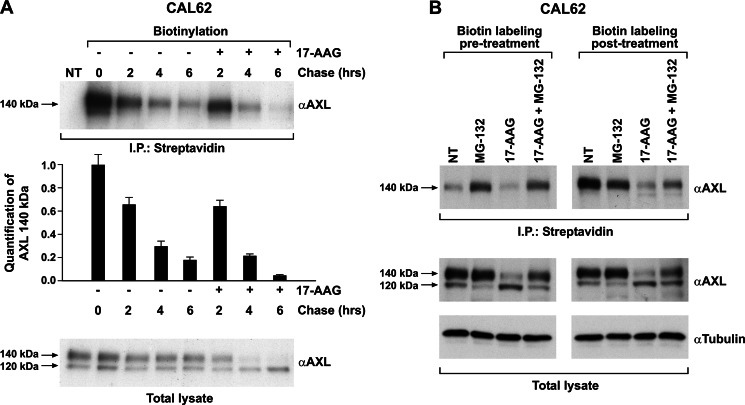
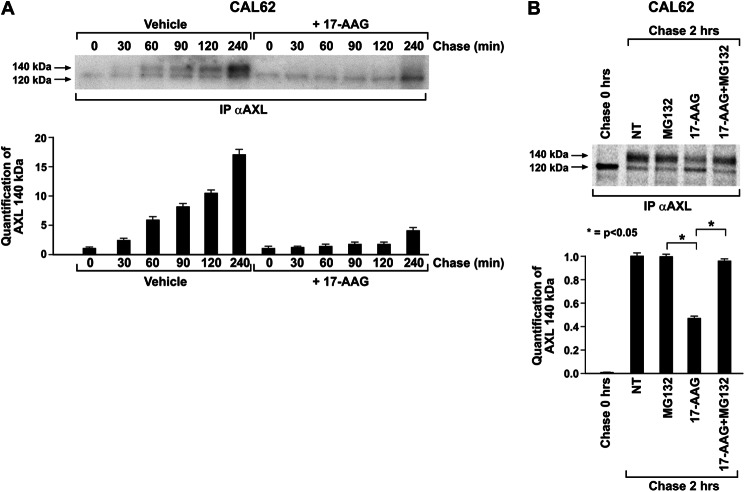
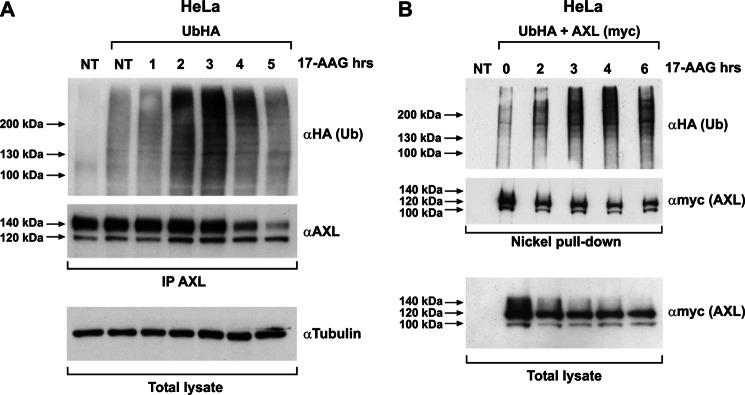
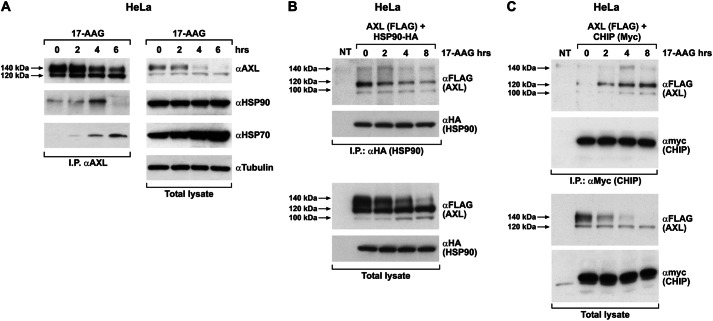
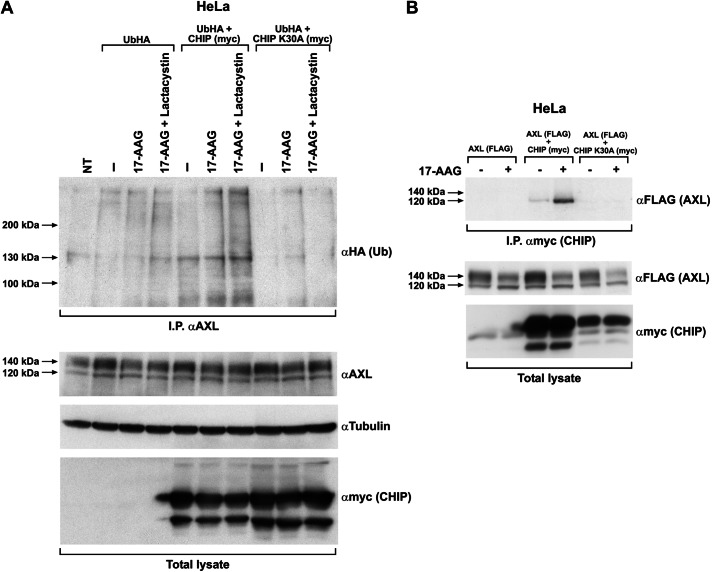
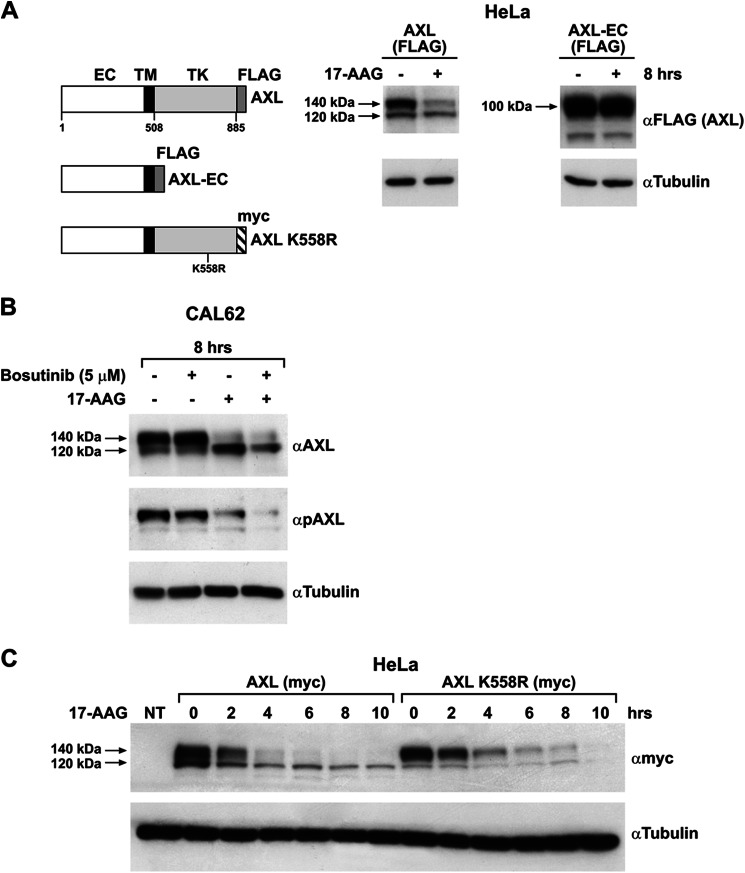
The receptor tyrosine kinase AXL is overexpressed in many cancer types including thyroid carcinomas and has well established roles in tumor formation and progression. Proper folding, maturation, and activity of several oncogenic receptor tyrosine kinases require HSP90 chaperoning. HSP90 inhibition by the antibiotic geldanamycin or its derivative 17-allylamino-17-demethoxygeldanamycin (17-AAG) causes destabilization of its client proteins. Here we show that AXL is a novel client protein of HSP90. 17-AAG induced a time- and dose-dependent down-regulation of endogenous or ectopically expressed AXL protein, thereby inhibiting AXL-mediated signaling and biological activity. 17-AAG-induced AXL down-regulation specifically affected fully glycosylated mature receptor present on cell membrane. By using biotin and [(35)S]methionine labeling, we showed that 17-AAG caused depletion of membrane-localized AXL by mediating its degradation in the intracellular compartment, thus restricting its exposure on the cell surface. 17-AAG induced AXL polyubiquitination and subsequent proteasomal degradation; under basal conditions, AXL co-immunoprecipitated with HSP90. Upon 17-AAG treatment, AXL associated with the co-chaperone HSP70 and the ubiquitin E3 ligase carboxyl terminus of HSC70-interacting protein (CHIP). Overexpression of CHIP, but not of the inactive mutant CHIP K30A, induced accumulation of AXL polyubiquitinated species upon 17-AAG treatment. The sensitivity of AXL to 17-AAG required its intracellular domain because an AXL intracellular domain-deleted mutant was insensitive to the compound. Active AXL and kinase-dead AXL were similarly sensitive to 17-AAG, implying that 17-AAG sensitivity does not require receptor phosphorylation. Overall our data elucidate the molecular basis of AXL down-regulation by HSP90 inhibitors and suggest that HSP90 inhibition in anticancer therapy can exert its effect through inhibition of multiple kinases including AXL.
Keywords: 17-AAG/Geldanamycin; AXL Receptor Tyrosine Kinase; CHIP E3 Ligase; E3 Ubiquitin Ligase; HSP90 Inhibition; Hsp90; Proteasome; Receptor Tyrosine Kinase; Ubiquitination.
Figures
References
-
- O'Bryan J. P., Frye R. A., Cogswell P. C., Neubauer A., Kitch B., Prokop C., Espinosa R., 3rd, Le Beau M. M., Earp H. S., Liu E. T. (1991) Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 11, 5016–5031 - PMC - PubMed
-
- Stitt T. N., Conn G., Gore M., Lai C., Bruno J., Radziejewski C., Mattsson K., Fisher J., Gies D. R., Jones P. F., Masiakowski P., Ryan T. E., Tobkes N. J., Chen D. H., DiStefano P. S., Long G. L., Basilico C., Goldfarb M. P., Lemke G., Glass D. J., Yancopoulos G. D. (1995) The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 80, 661–670 - PubMed
-
- Holland S. J., Pan A., Franci C., Hu Y., Chang B., Li W., Duan M., Torneros A., Yu J., Heckrodt T. J., Zhang J., Ding P., Apatira A., Chua J., Brandt R., Pine P., Goff D., Singh R., Payan D. G., Hitoshi Y. (2010) R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 70, 1544–1554 - PubMed
-
- Zhang Y. X., Knyazev P. G., Cheburkin Y. V., Sharma K., Knyazev Y. P., Orfi L., Szabadkai I., Daub H., Kéri G., Ullrich A. (2008) AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 68, 1905–1915 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
