

Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses
- PMID: 23630620
- PMCID: PMC3632529
- DOI: 10.1371/journal.pone.0061950
Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses
Erratum in
- PLoS One. 2013;8(6). doi:10.1371/annotation/68f77773-a2a0-4bfe-b5e6-950dc30b79f9
Abstract
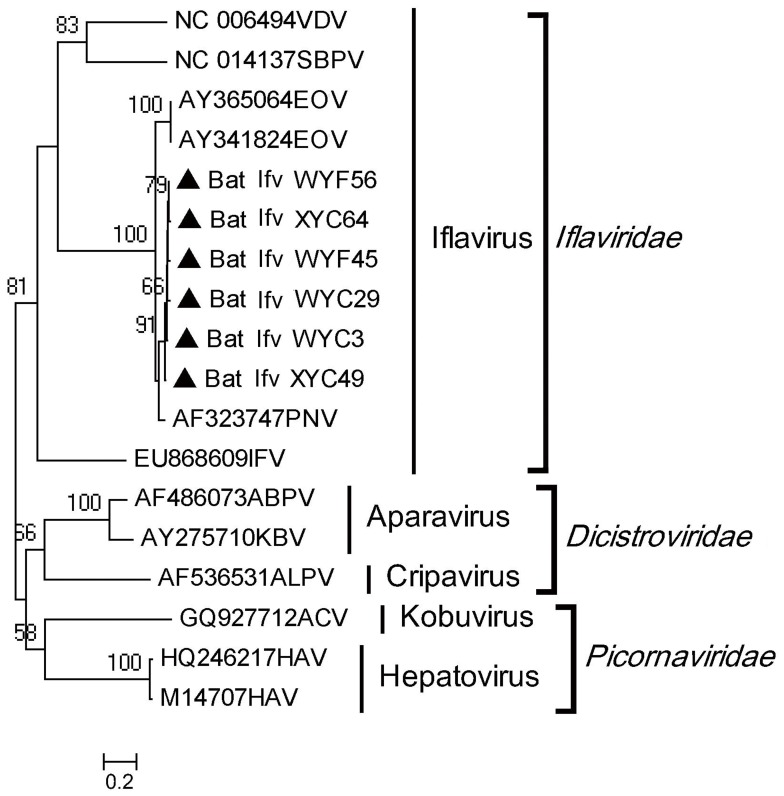
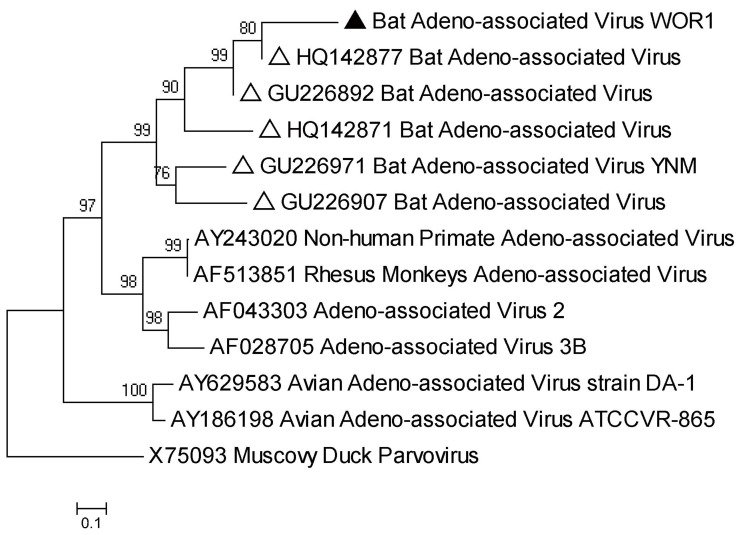
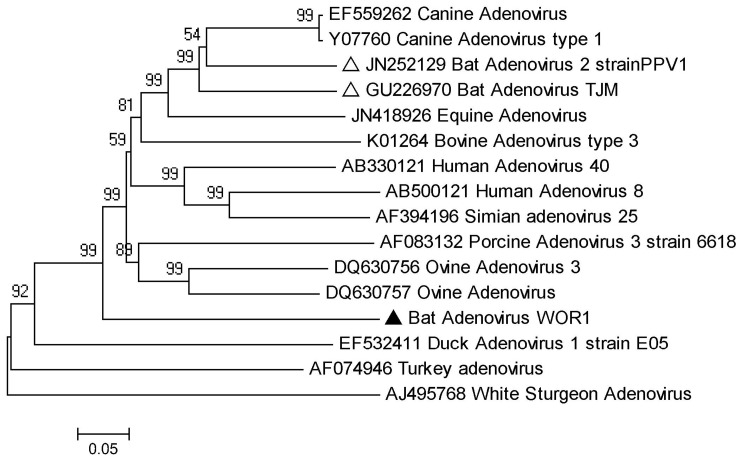
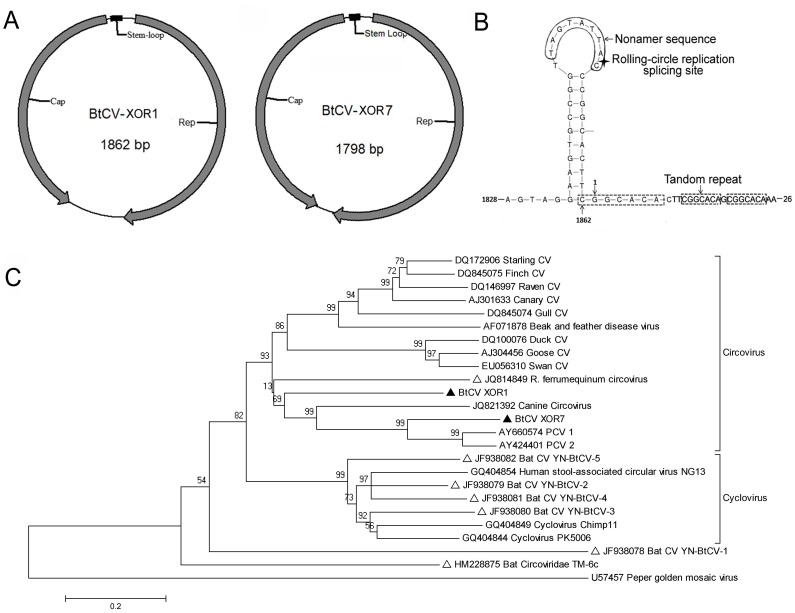
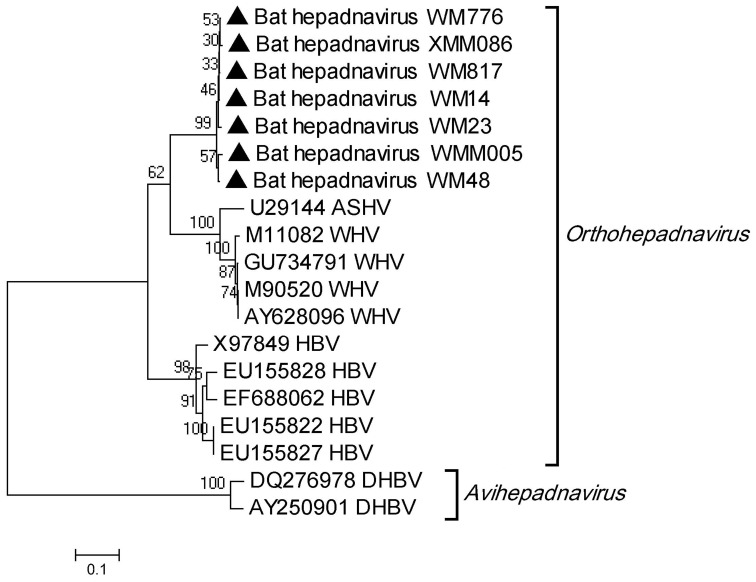
Bats are reservoir animals harboring many important pathogenic viruses and with the capability of transmitting these to humans and other animals. To establish an effective surveillance to monitor transboundary spread of bat viruses between Myanmar and China, complete organs from the thorax and abdomen from 853 bats of six species from two Myanmar counties close to Yunnan province, China, were collected and tested for their virome through metagenomics by Solexa sequencing and bioinformatic analysis. In total, 3,742,314 reads of 114 bases were generated, and over 86% were assembled into 1,649,512 contigs with an average length of 114 bp, of which 26,698 (2%) contigs were recognizable viral sequences belonging to 24 viral families. Of the viral contigs 45% (12,086/26,698) were related to vertebrate viruses, 28% (7,443/26,698) to insect viruses, 27% (7,074/26,698) to phages and 95 contigs to plant viruses. The metagenomic results were confirmed by PCR of selected viruses in all bat samples followed by phylogenetic analysis, which has led to the discovery of some novel bat viruses of the genera Mamastrovirus, Bocavirus, Circovirus, Iflavirus and Orthohepadnavirus and to their prevalence rates in two bat species. In conclusion, the present study aims to present the bat virome in Myanmar, and the results obtained further expand the spectrum of viruses harbored by bats.
Conflict of interest statement
Figures


References
-
- Simmons NB (2005) Order Chiroptera. In: Wilson DE, Reeder DM, editor. Mammal species of the world: a taxonomic and geographic reference (3rd ed). Baltimore: Johns Hopkins University Press, 312–529.
-
- Halpin K, Young PL, Field HE, Mackenzie JS (2000) Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. J Gen Virol 81: 1927–1932. - PubMed
-
- Kuzmin IV, Mayer AE, Niezgoda M, Markotter W, Agwanda B, et al. (2010) Shimoni bat virus, a new representative of the Lyssavirus genus. Virus Res 149: 197–210. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
