

Peptide crystal simulations reveal hidden dynamics
- PMID: 23631449
- PMCID: PMC3668435
- DOI: 10.1021/ja401382y
Peptide crystal simulations reveal hidden dynamics
Abstract
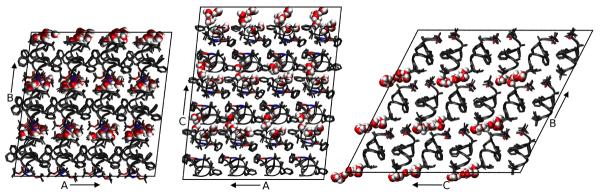
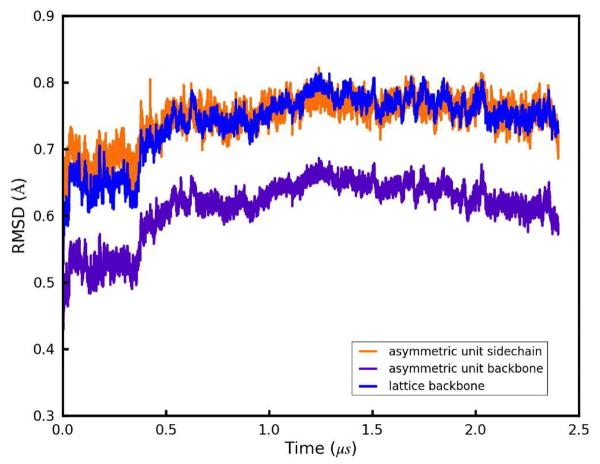
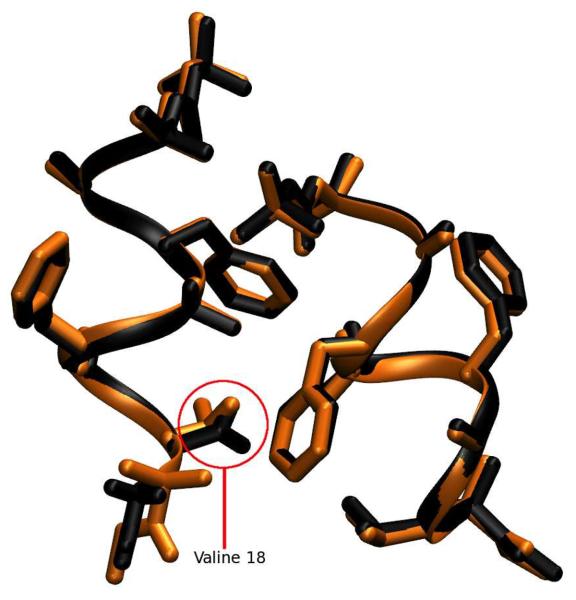
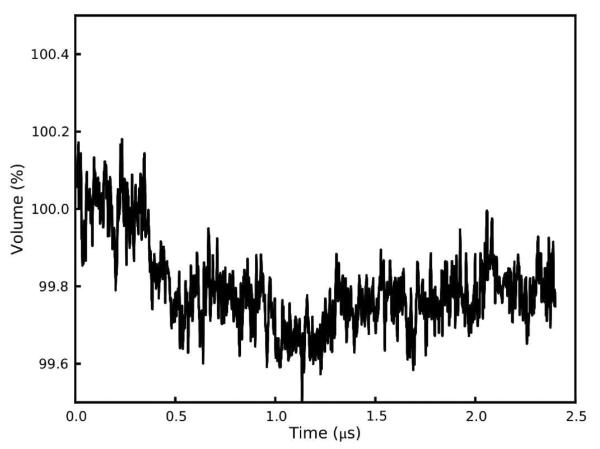
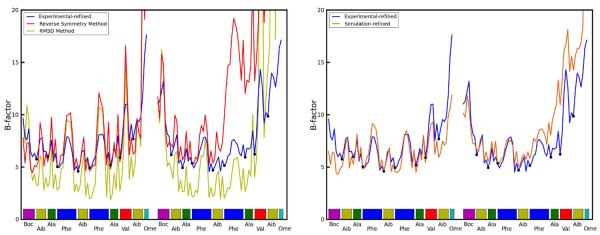
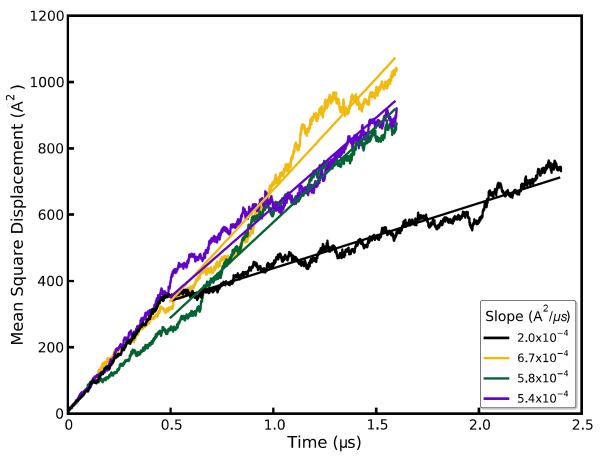
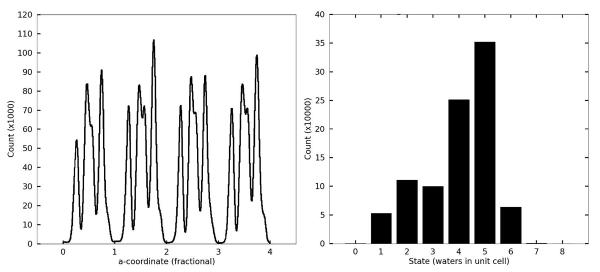
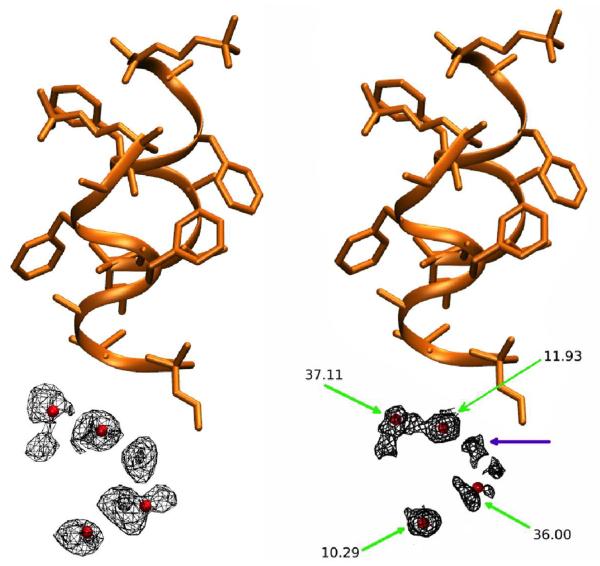
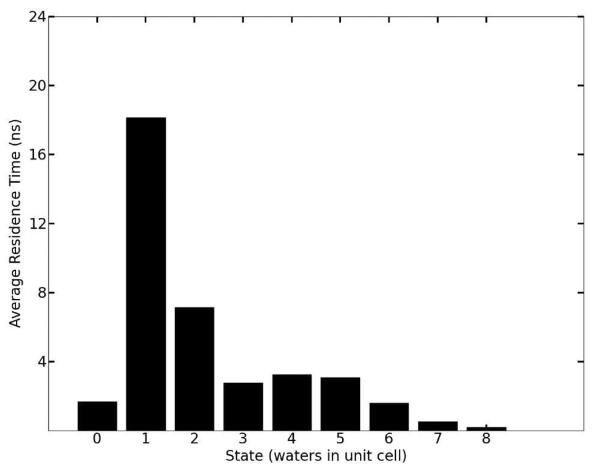
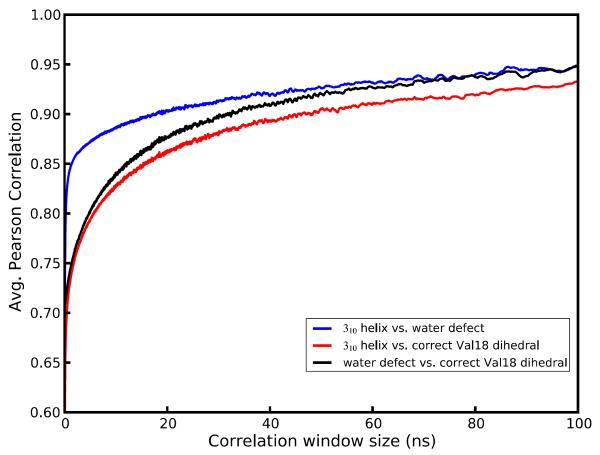
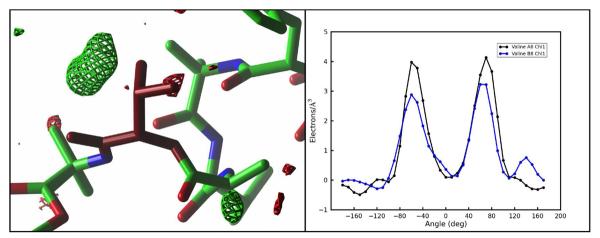
Molecular dynamics simulations of biomolecular crystals at atomic resolution have the potential to recover information on dynamics and heterogeneity hidden in X-ray diffraction data. We present here 9.6 μs of dynamics in a small helical peptide crystal with 36 independent copies of the unit cell. The average simulation structure agrees with experiment to within 0.28 Å backbone and 0.42 Å all-atom RMSD; a model refined against the average simulation density agrees with the experimental structure to within 0.20 Å backbone and 0.33 Å all-atom RMSD. The R-factor between the experimental structure factors and those derived from this unrestrained simulation is 23% to 1.0 Å resolution. The B-factors for most heavy atoms agree well with experiment (Pearson correlation of 0.90), but B-factors obtained by refinement against the average simulation density underestimate the coordinate fluctuations in the underlying simulation where the simulation samples alternate conformations. A dynamic flow of water molecules through channels within the crystal lattice is observed, yet the average water density is in remarkable agreement with experiment. A minor population of unit cells is characterized by reduced water content, 310 helical propensity and a gauche(-) side-chain rotamer for one of the valine residues. Careful examination of the experimental data suggests that transitions of the helices are a simulation artifact, although there is indeed evidence for alternate valine conformers and variable water content. This study highlights the potential for crystal simulations to detect dynamics and heterogeneity in experimental diffraction data as well as to validate computational chemistry methods.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
