

RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species
- PMID: 23633517
- PMCID: PMC3765041
- DOI: 10.1093/carcin/bgt143
RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species
Abstract
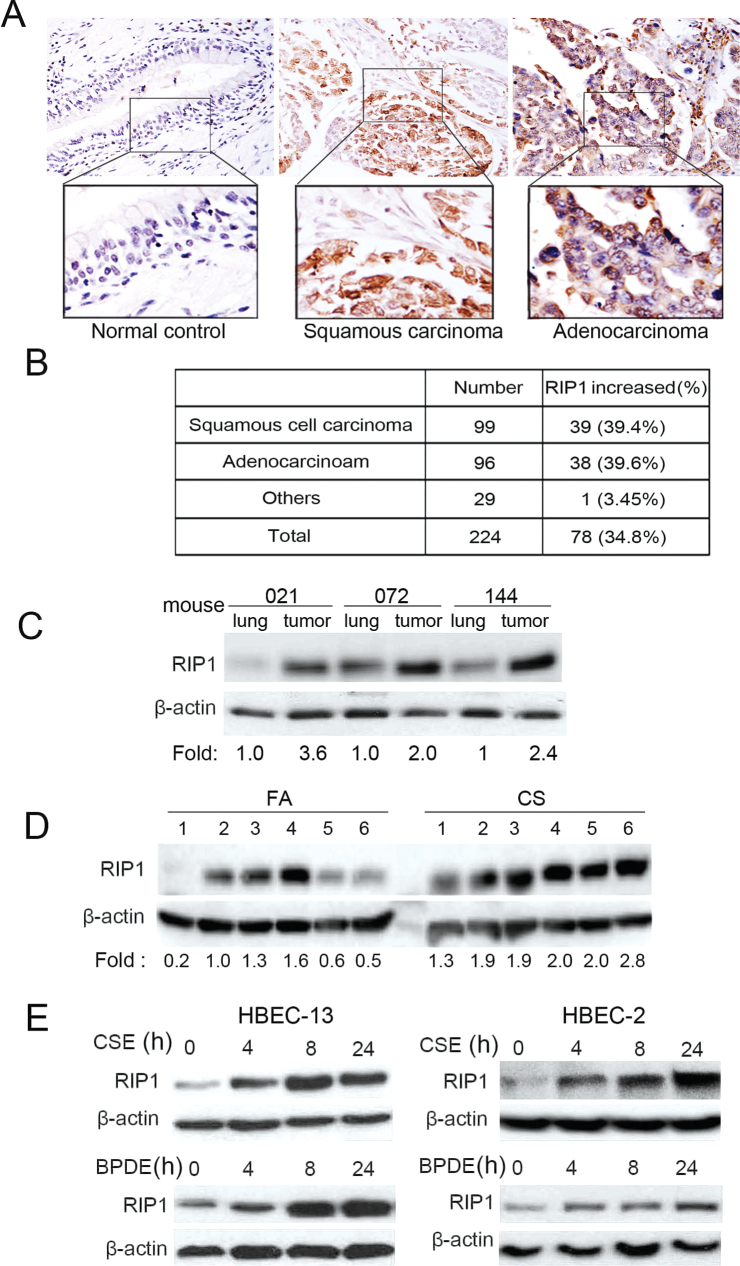
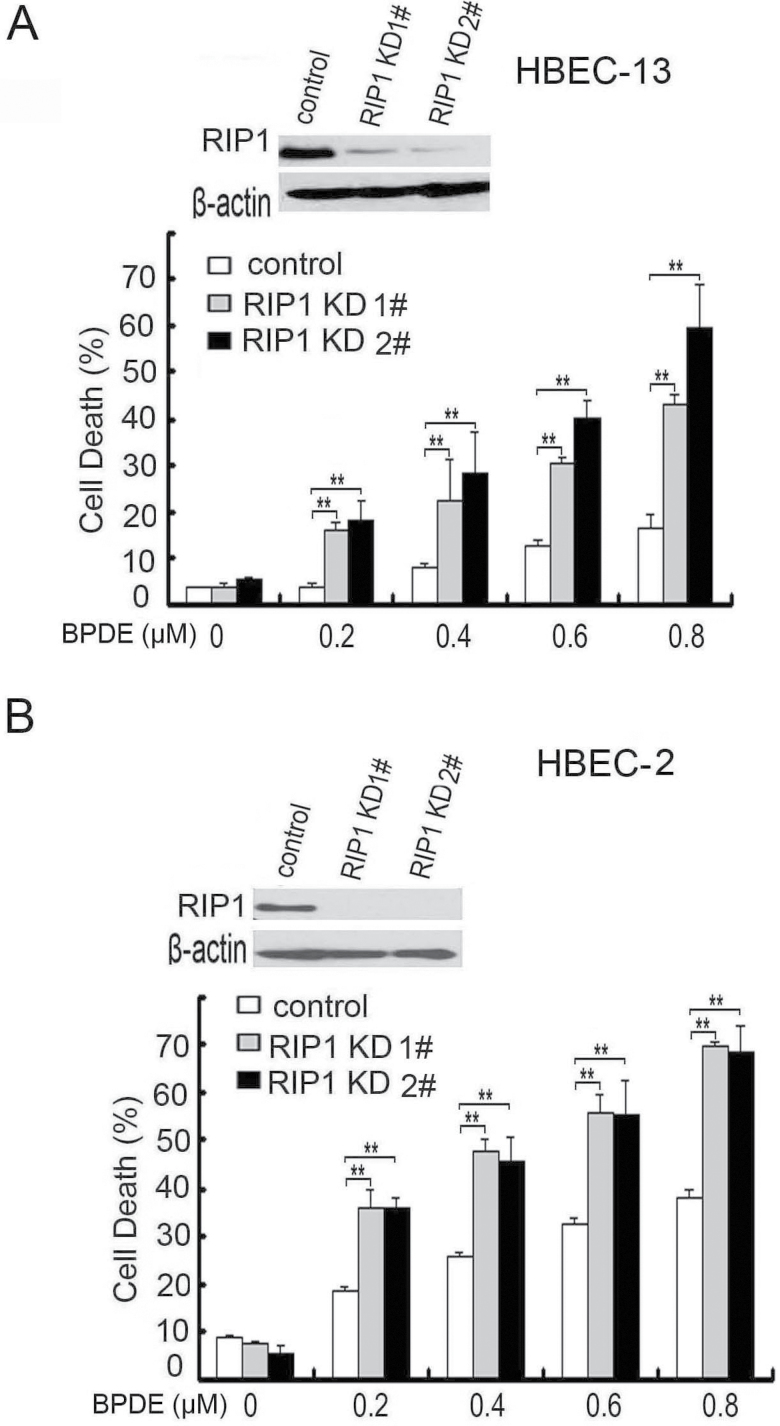
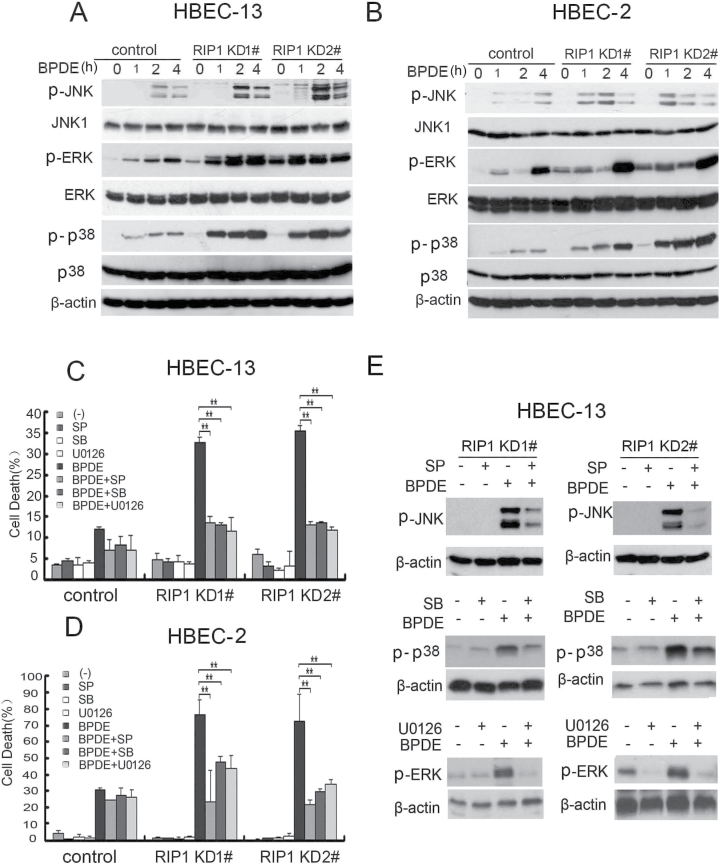
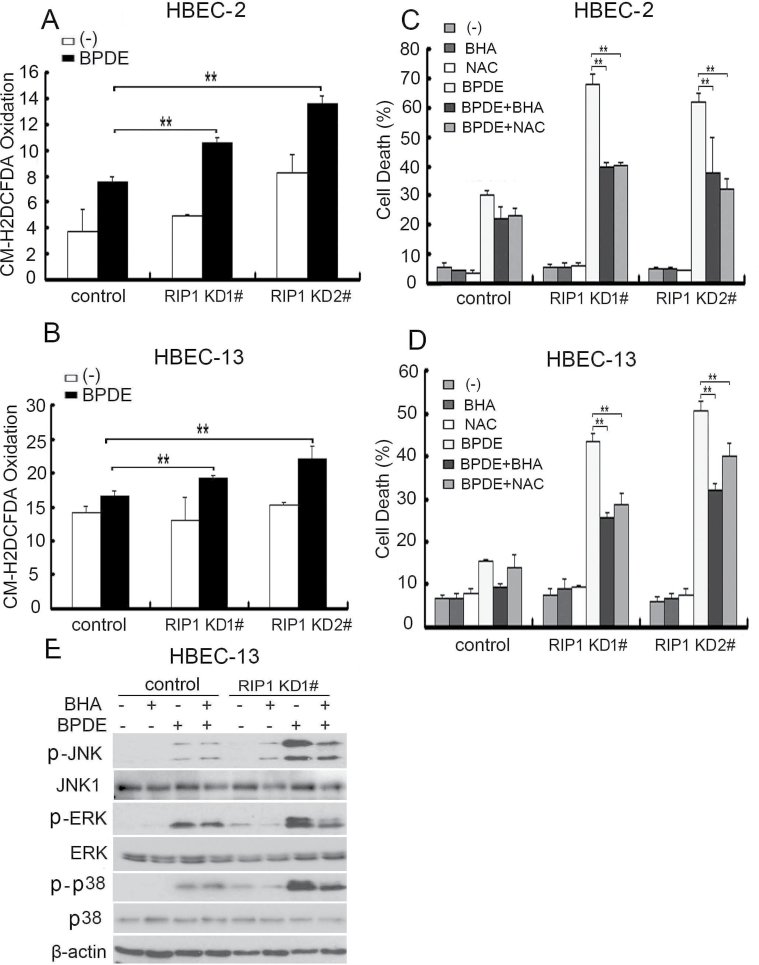
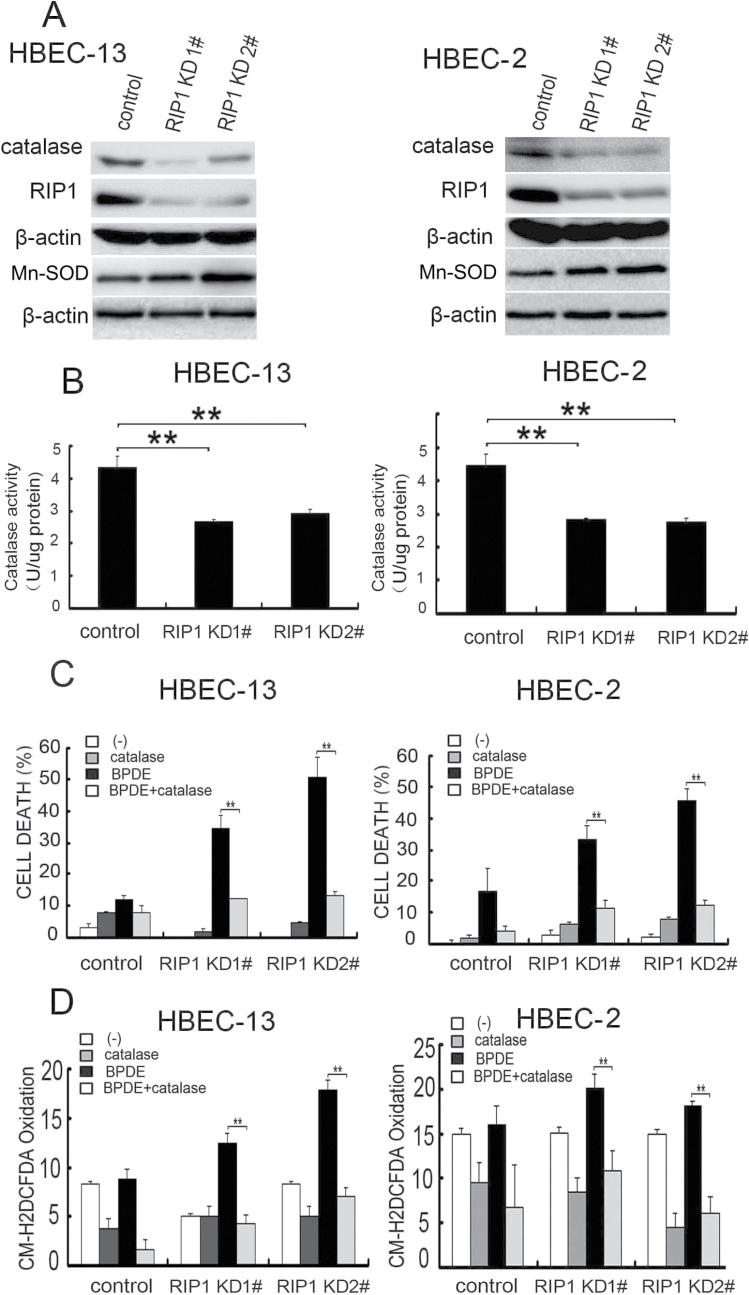
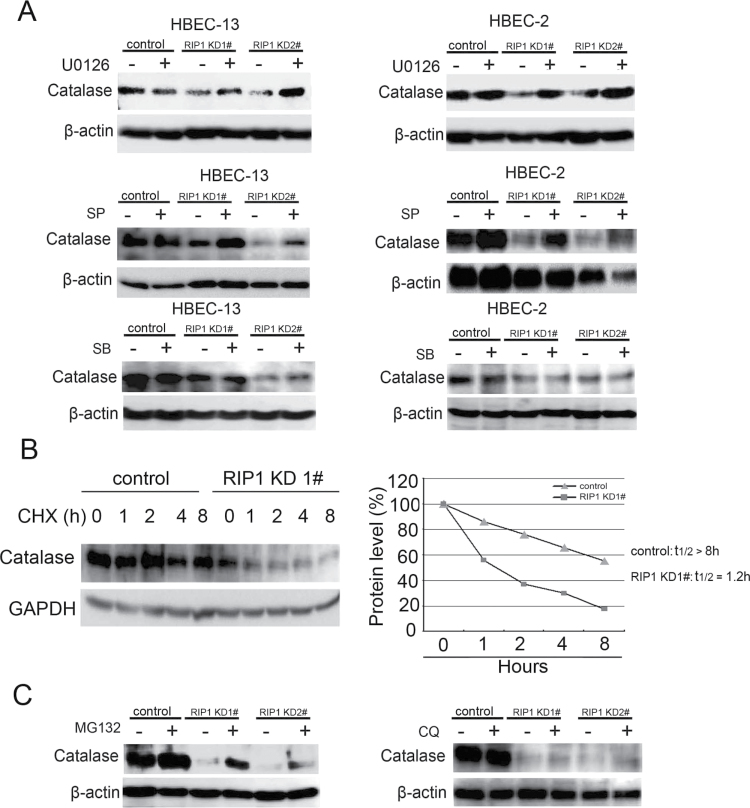
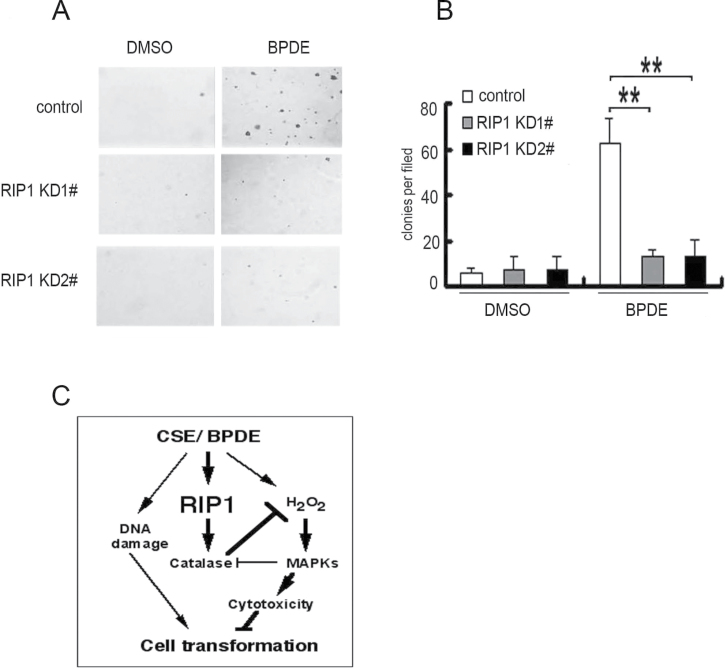
Cell survival signaling is important for the malignant phenotypes of cancer cells. Although the role of receptor-interacting protein 1 (RIP1) in cell survival signaling is well documented, whether RIP1 is directly involved in cancer development has never been studied. In this report, we found that RIP1 expression is substantially increased in human non-small cell lung cancer and mouse lung tumor tissues. RIP1 expression was remarkably increased in cigarette smoke-exposed mouse lung. In human bronchial epithelial cells (HBECs), RIP1 was significantly induced by cigarette smoke extract or benzo[a]pyrene diol epoxide (BPDE), the active form of the tobacco-specific carcinogen benzo(a)pyrene. In RIP1 knockdown HBECs, BPDE-induced cytotoxicity was significantly increased, which was associated with induction of cellular reactive oxygen species (ROS) and activation of mitogen-activated protein kinases (MAPKs), including c-jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK) and p38. Scavenging ROS suppressed BPDE-induced MAPK activation and inhibiting ROS or MAPKs substantially blocked BPDE-induced cytotoxicity, suggesting ROS-mediated MAPK activation is involved in BPDE-induced cell death. The ROS-reducing enzyme catalase is destabilized in an ERK- and JNK-dependent manner in RIP1 knockdown HBECs and application of catalase effectively blocked BPDE-induced ROS accumulation and cytotoxicity. Importantly, BPDE-induced transformation of HBECs was significantly reduced when RIP1 expression was suppressed. Altogether, these results strongly suggest an oncogenic role for RIP1, which promotes malignant transformation through protecting DNA-damaged cells against carcinogen-induced cytotoxicity associated with excessive ROS production.
Figures
Similar articles
-
Receptor-interacting protein 1 increases chemoresistance by maintaining inhibitor of apoptosis protein levels and reducing reactive oxygen species through a microRNA-146a-mediated catalase pathway.J Biol Chem. 2014 Feb 28;289(9):5654-63. doi: 10.1074/jbc.M113.526152. Epub 2014 Jan 14. J Biol Chem. 2014. PMID: 24425875 Free PMC article.
-
Cytosolic calcium mediates RIP1/RIP3 complex-dependent necroptosis through JNK activation and mitochondrial ROS production in human colon cancer cells.Free Radic Biol Med. 2017 Jul;108:433-444. doi: 10.1016/j.freeradbiomed.2017.04.010. Epub 2017 Apr 14. Free Radic Biol Med. 2017. PMID: 28414098
-
MUC1 contributes to BPDE-induced human bronchial epithelial cell transformation through facilitating EGFR activation.PLoS One. 2012;7(3):e33846. doi: 10.1371/journal.pone.0033846. Epub 2012 Mar 22. PLoS One. 2012. PMID: 22457794 Free PMC article.
-
The critical DNA damage by benzo(a)pyrene in lung tissues of smokers and approaches to preventing its formation.Toxicol Lett. 2010 Sep 15;198(1):63-8. doi: 10.1016/j.toxlet.2010.04.009. Epub 2010 Apr 24. Toxicol Lett. 2010. PMID: 20399842 Review.
-
The role of the kinases RIP1 and RIP3 in TNF-induced necrosis.Sci Signal. 2010 Mar 30;3(115):re4. doi: 10.1126/scisignal.3115re4. Sci Signal. 2010. PMID: 20354226 Review.
Cited by
-
The potential role of necroptosis in clinical diseases (Review).Int J Mol Med. 2021 May;47(5):89. doi: 10.3892/ijmm.2021.4922. Epub 2021 Mar 31. Int J Mol Med. 2021. PMID: 33786617 Free PMC article. Review.
-
Immunonutrition, Metabolism, and Programmed Cell Death in Lung Cancer: Translating Bench to Bedside.Biology (Basel). 2024 Jun 4;13(6):409. doi: 10.3390/biology13060409. Biology (Basel). 2024. PMID: 38927289 Free PMC article. Review.
-
Integrated analysis of necroptosis-related genes for evaluating immune infiltration and colon cancer prognosis.Front Immunol. 2022 Dec 22;13:1085038. doi: 10.3389/fimmu.2022.1085038. eCollection 2022. Front Immunol. 2022. PMID: 36618366 Free PMC article.
-
MUC1 in macrophage: contributions to cigarette smoke-induced lung cancer.Cancer Res. 2014 Jan 15;74(2):460-70. doi: 10.1158/0008-5472.CAN-13-1713. Epub 2013 Nov 26. Cancer Res. 2014. PMID: 24282280 Free PMC article.
-
Vasorin/ATIA Promotes Cigarette Smoke-Induced Transformation of Human Bronchial Epithelial Cells by Suppressing Autophagy-Mediated Apoptosis.Transl Oncol. 2020 Jan;13(1):32-41. doi: 10.1016/j.tranon.2019.09.001. Epub 2019 Nov 21. Transl Oncol. 2020. PMID: 31760267 Free PMC article.
References
-
- Hanahan D., et al. (2011). Hallmarks of cancer: the next generation. Cell, 144, 646–674 - PubMed
-
- Festjens N., et al. (2007). RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ., 14, 400–410 - PubMed
-
- Meylan E., et al. (2004). RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol., 5, 503–507 - PubMed
-
- Kelliher M.A., et al. (1998). The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity, 8, 297–303 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
