

HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1
- PMID: 23633945
- PMCID: PMC3635988
- DOI: 10.1371/journal.ppat.1003248
HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1
Abstract
Upon recognition of viral components by pattern recognition receptors, such as the toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I)-like helicases, cells are activated to produce type I interferon (IFN) and proinflammatory cytokines. These pathways are tightly regulated by the host to prevent an inappropriate cellular response, but viruses can modulate these pathways to proliferate and spread. In this study, we revealed a novel mechanism in which hepatitis C virus (HCV) evades the immune surveillance system to proliferate by activating microRNA-21 (miR-21). We demonstrated that HCV infection upregulates miR-21, which in turn suppresses HCV-triggered type I IFN production, thus promoting HCV replication. Furthermore, we demonstrated that miR-21 targets two important factors in the TLR signaling pathway, myeloid differentiation factor 88 (MyD88) and interleukin-1 receptor-associated kinase 1 (IRAK1), which are involved in HCV-induced type I IFN production. HCV-mediated activation of miR-21 expression requires viral proteins and several signaling components. Moreover, we identified a transcription factor, activating protein-1 (AP-1), which is partly responsible for miR-21 induction in response to HCV infection through PKCε/JNK/c-Jun and PKCα/ERK/c-Fos cascades. Taken together, our results indicate that miR-21 is upregulated during HCV infection and negatively regulates IFN-α signaling through MyD88 and IRAK1 and may be a potential therapeutic target for antiviral intervention.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
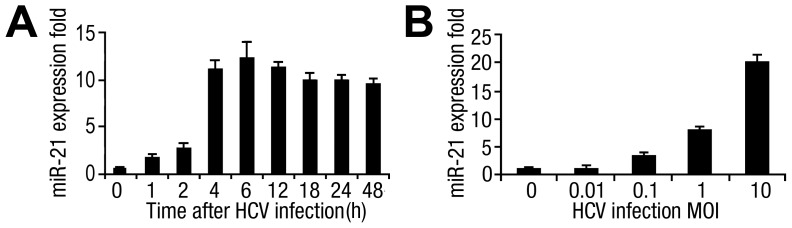
 SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments.
SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments.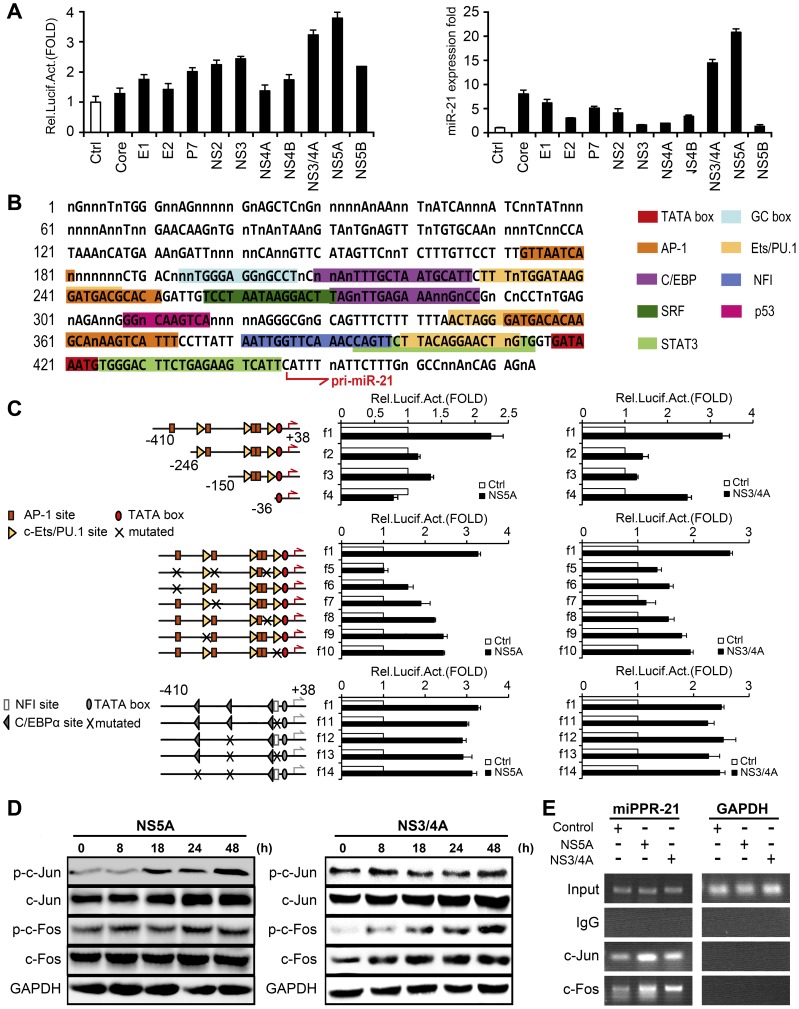
 SD from three experiments. (D) Huh7 cells were transfected with pCMV-NS5A or pCMV-NS3/4A at different time intervals, respectively. The phosphorylation and total protein levels of c-Jun and c-Fos were determined by Western blot. (E) AP-1 binding sites were determined by ChIP assays. All experiments were repeated at least three times with similar results.
SD from three experiments. (D) Huh7 cells were transfected with pCMV-NS5A or pCMV-NS3/4A at different time intervals, respectively. The phosphorylation and total protein levels of c-Jun and c-Fos were determined by Western blot. (E) AP-1 binding sites were determined by ChIP assays. All experiments were repeated at least three times with similar results.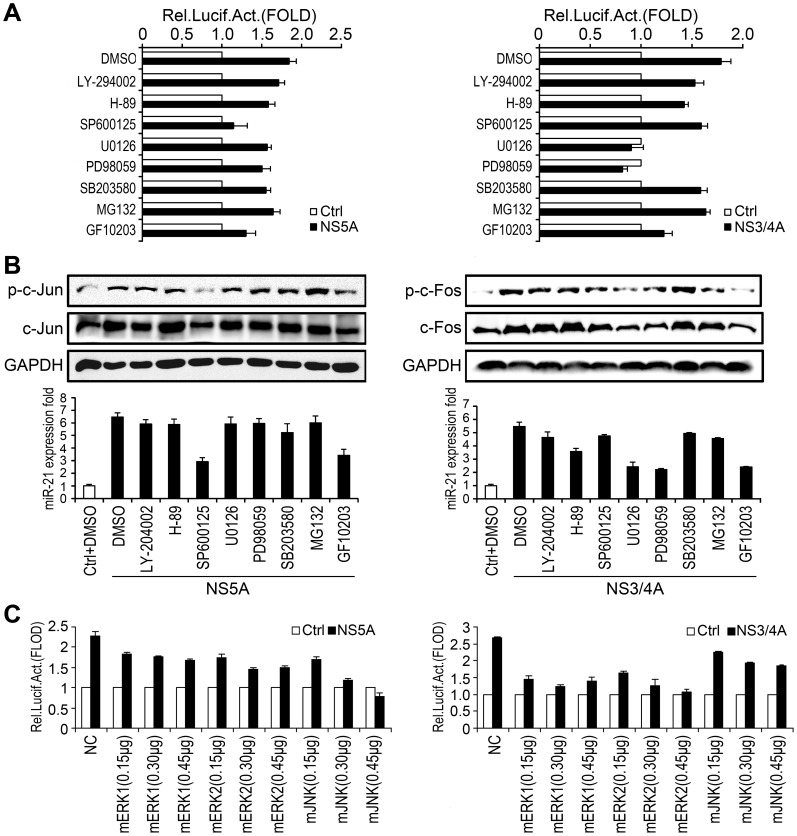
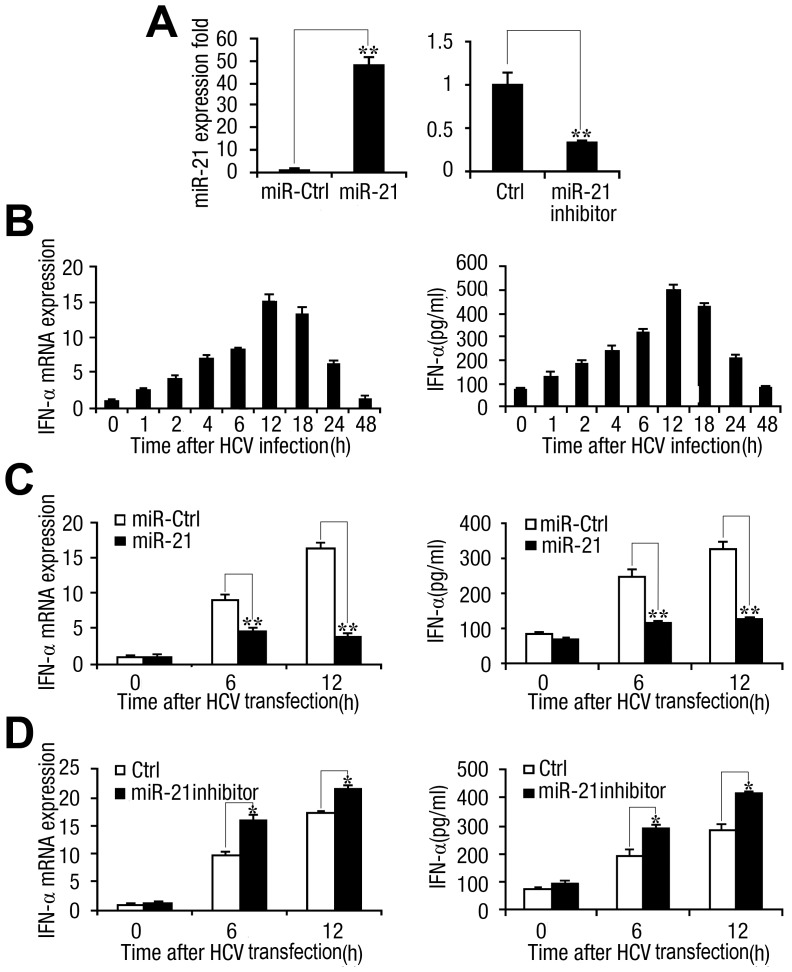
 SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.
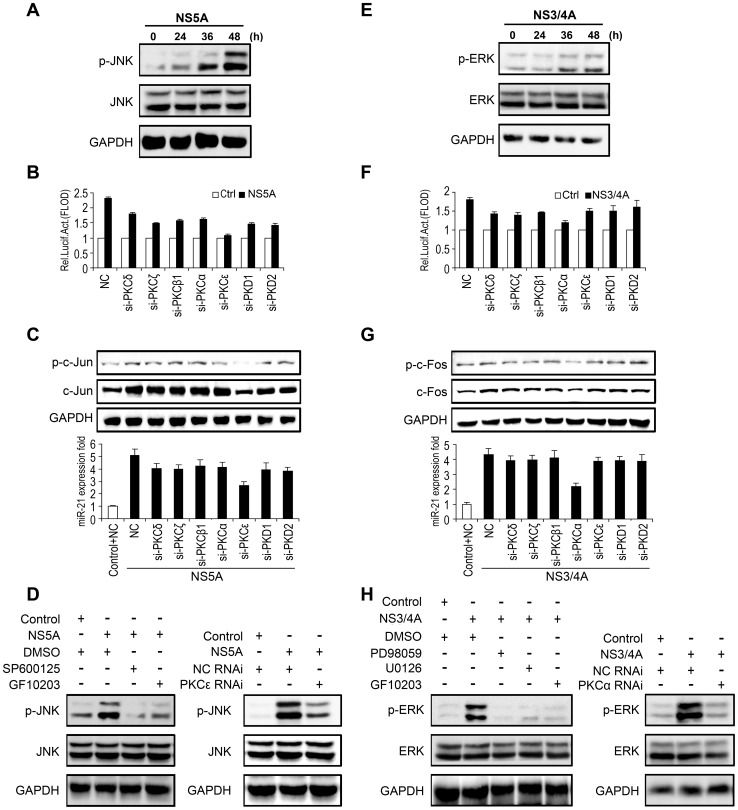
SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.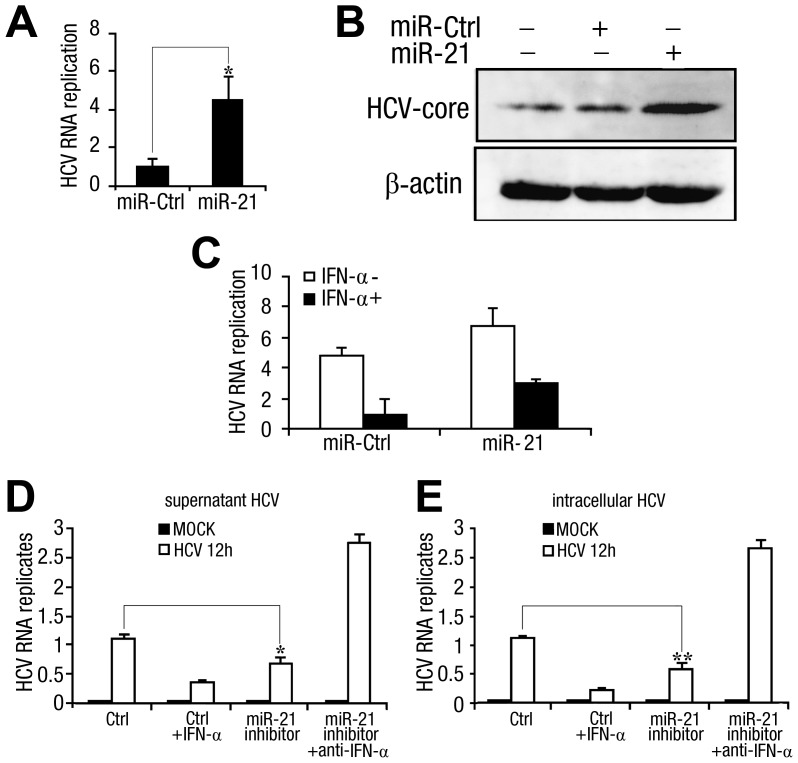
 SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.
SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.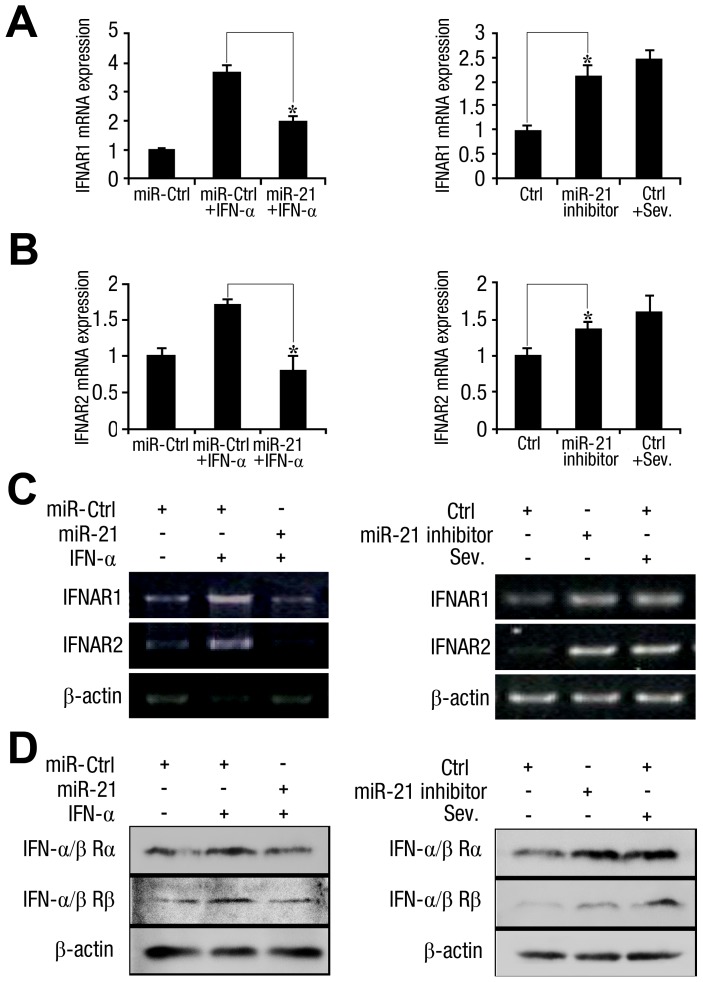
 SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.
SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.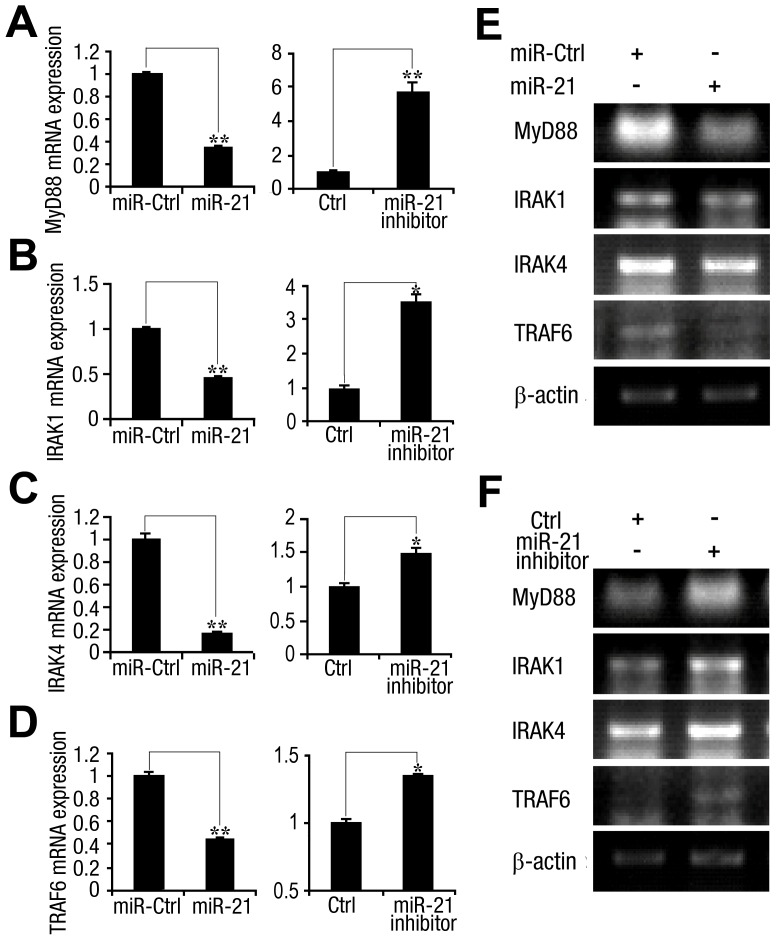
 SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.
SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.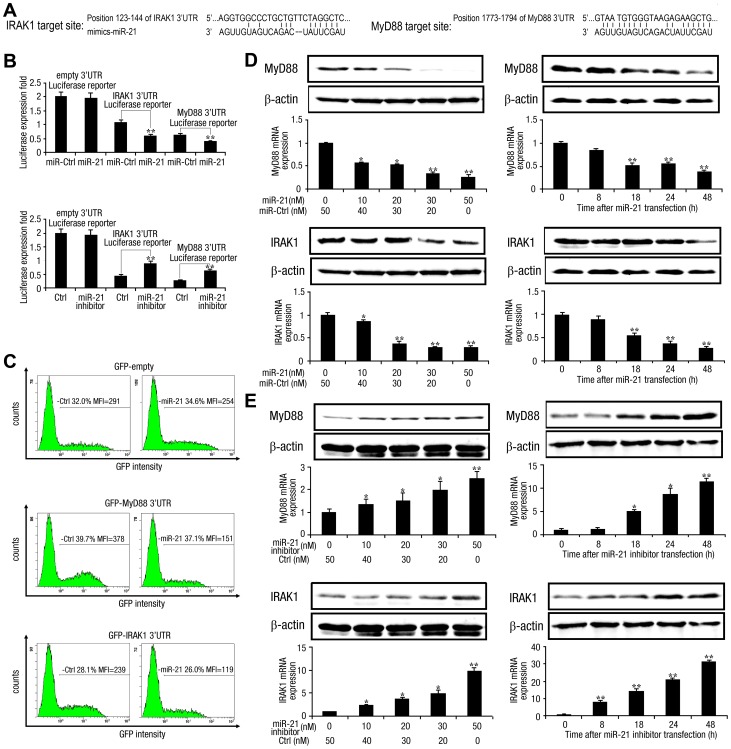
 SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.
SD (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.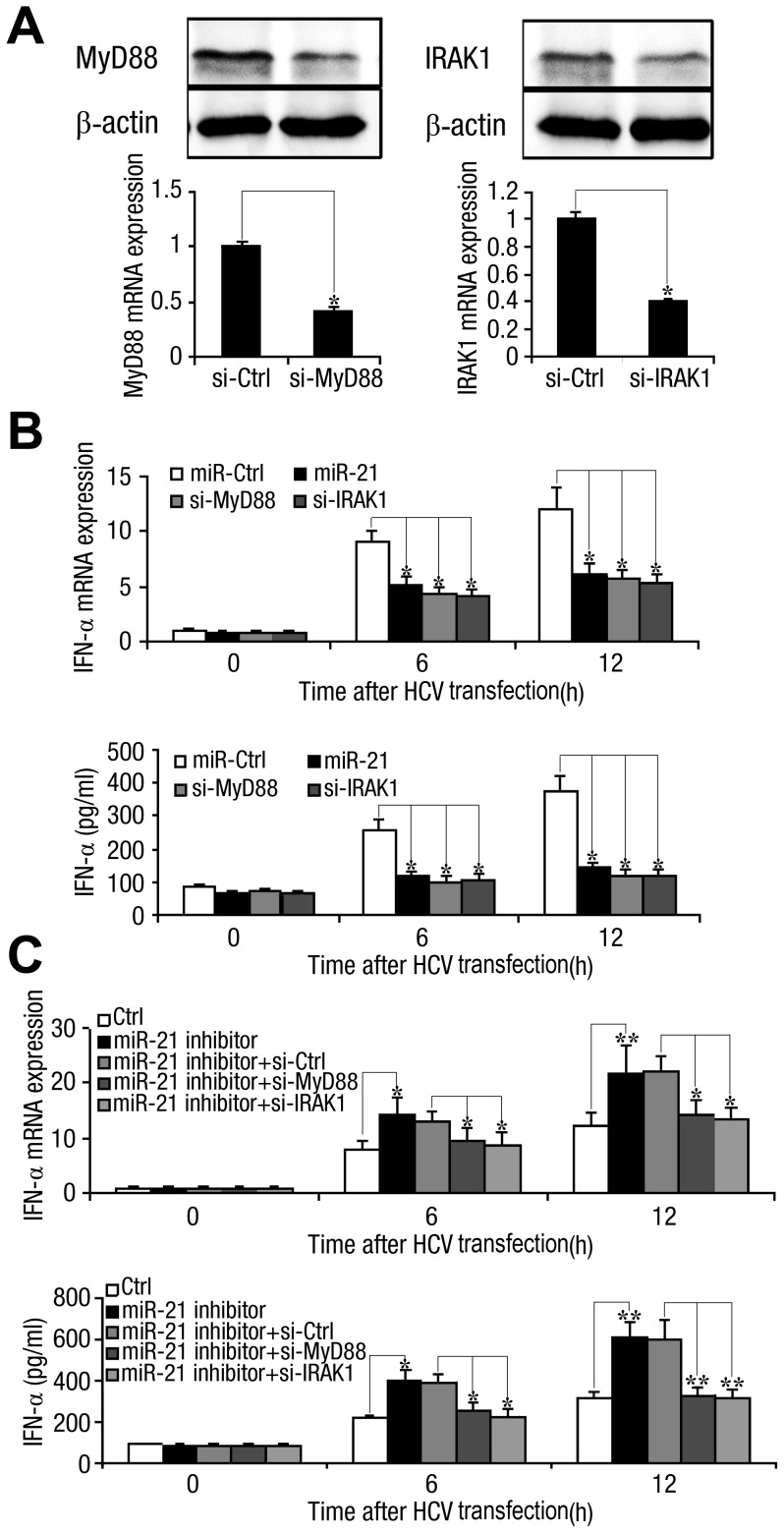
 SD (n = 3) of one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.
SD (n = 3) of one representative experiment. Similar results were obtained in three independent experiments. **, p<0.01; *, p<0.05.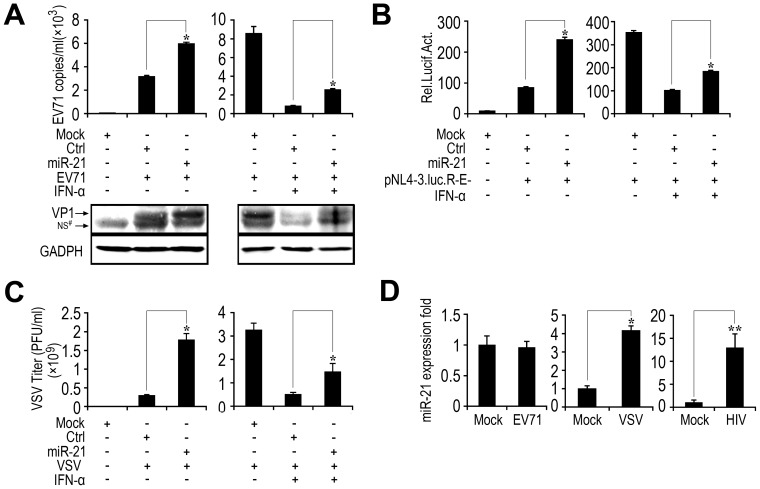
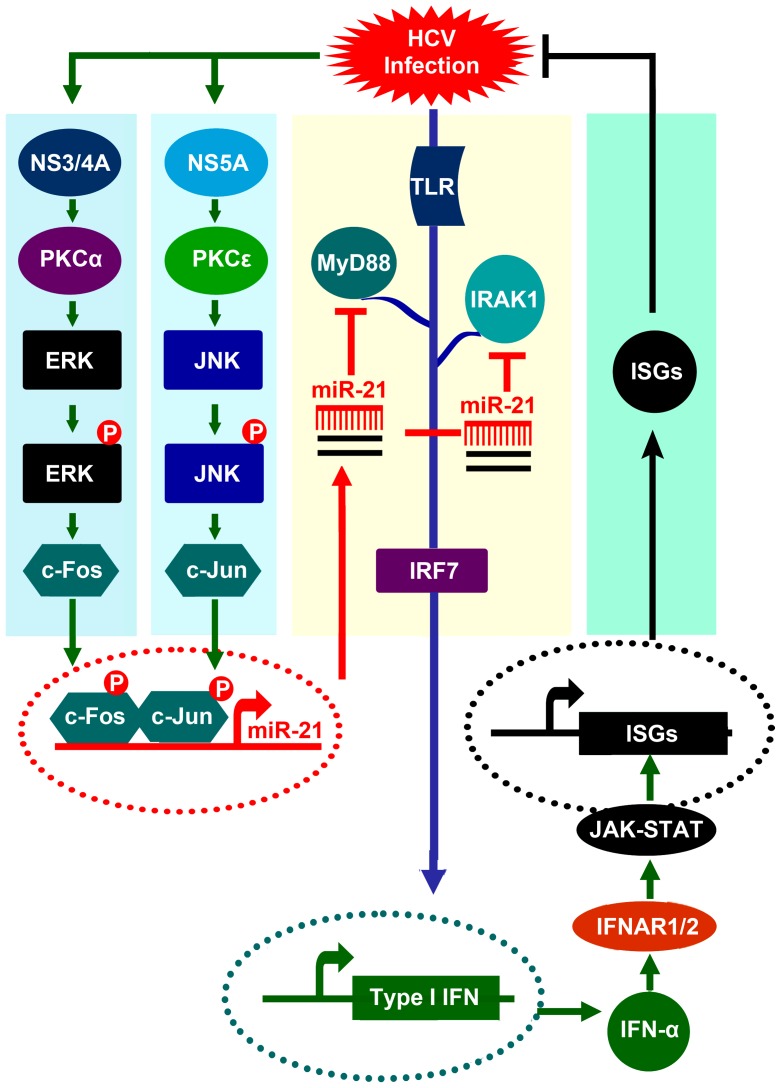
References
-
- Houghton M, Weiner A, Han J, Kuo G, Choo QL (1991) Molecular biology of the hepatitis C viruses: implications for diagnosis, development and control of viral disease. Hepatology 14: 381–388. - PubMed
-
- Robertson B, Myers G, Howard C, Brettin T, Bukh J, et al. (1998) Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch Virol 143: 2493–2503. - PubMed
-
- Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, et al. (1989) Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244: 359–362. - PubMed
-
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, et al. (2005) Complete replication of hepatitis C virus in cell culture. Science 309: 623–626. - PubMed
-
- Lauer GM, Walker BD (2001) Hepatitis C virus infection. N Engl J Med 345: 41–52. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
