

IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease
- PMID: 23633957
- PMCID: PMC3635973
- DOI: 10.1371/journal.ppat.1003330
IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease
Abstract
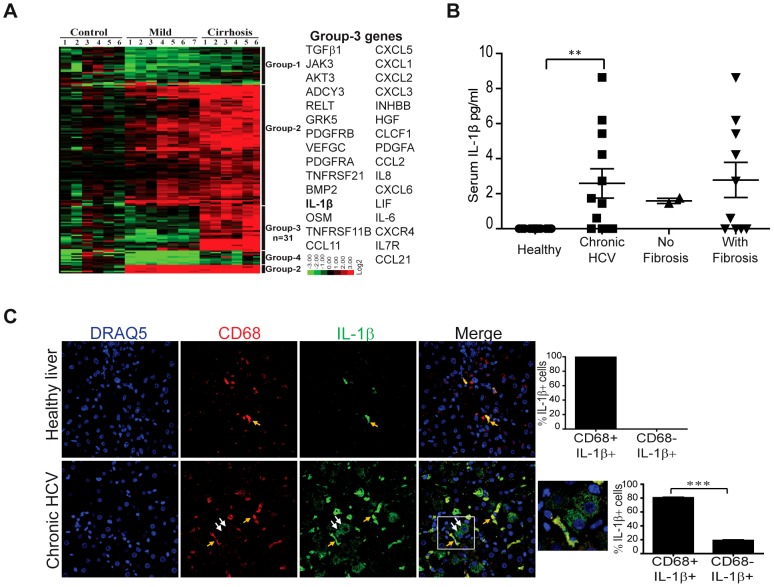
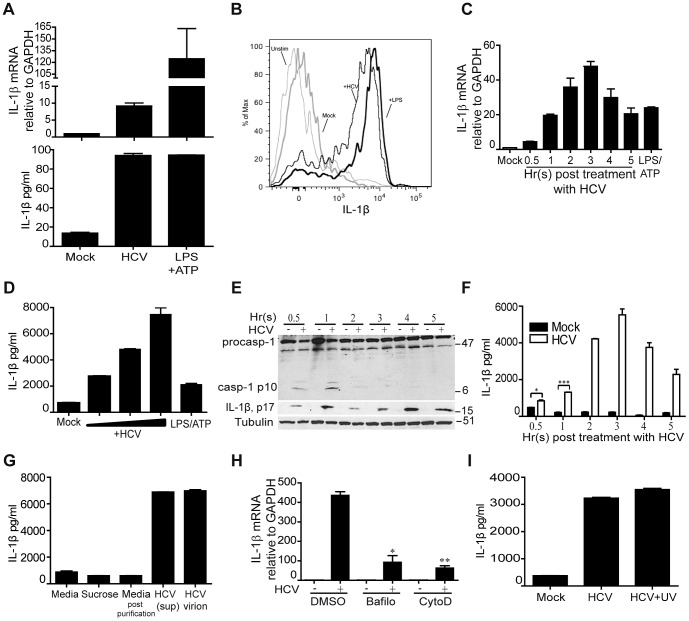
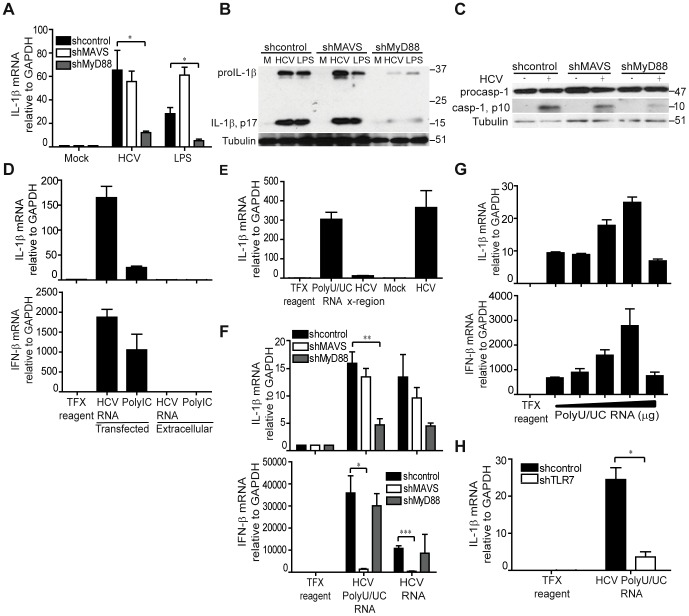
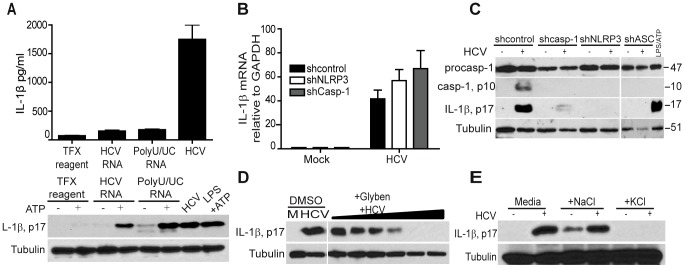
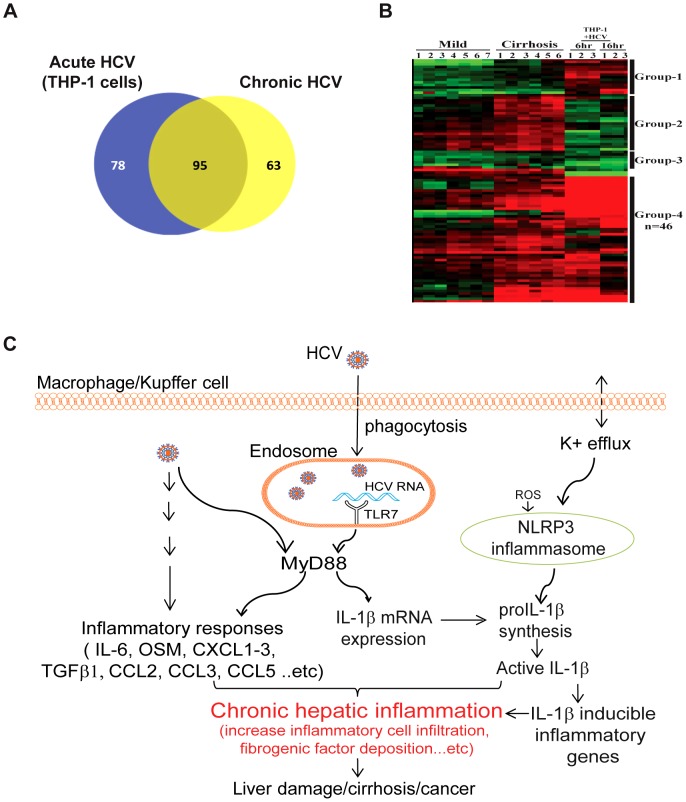
Chronic hepatitis C virus (HCV) infection is a leading cause of liver disease. Liver inflammation underlies infection-induced fibrosis, cirrhosis and liver cancer but the processes that promote hepatic inflammation by HCV are not defined. We provide a systems biology analysis with multiple lines of evidence to indicate that interleukin-1β (IL-1β) production by intrahepatic macrophages confers liver inflammation through HCV-induced inflammasome signaling. Chronic hepatitis C patients exhibited elevated levels of serum IL-1β compared to healthy controls. Immunohistochemical analysis of healthy control and chronic hepatitis C liver sections revealed that Kupffer cells, resident hepatic macrophages, are the primary cellular source of hepatic IL-1β during HCV infection. Accordingly, we found that both blood monocyte-derived primary human macrophages, and Kupffer cells recovered from normal donor liver, produce IL-1β after HCV exposure. Using the THP-1 macrophage cell-culture model, we found that HCV drives a rapid but transient caspase-1 activation to stimulate IL-1β secretion. HCV can enter macrophages through non-CD81 mediated phagocytic uptake that is independent of productive infection. Viral RNA triggers MyD88-mediated TLR7 signaling to induce IL-1β mRNA expression. HCV uptake concomitantly induces a potassium efflux that activates the NLRP3 inflammasome for IL-1β processing and secretion. RNA sequencing analysis comparing THP1 cells and chronic hepatitis C patient liver demonstrates that viral engagement of the NLRP3 inflammasome stimulates IL-1β production to drive proinflammatory cytokine, chemokine, and immune-regulatory gene expression networks linked with HCV disease severity. These studies identify intrahepatic IL-1β production as a central feature of liver inflammation during HCV infection. Thus, strategies to suppress NLRP3 or IL-1β activity could offer therapeutic actions to reduce hepatic inflammation and mitigate disease.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation.PLoS Pathog. 2019 Feb 27;15(2):e1007593. doi: 10.1371/journal.ppat.1007593. eCollection 2019 Feb. PLoS Pathog. 2019. PMID: 30811485 Free PMC article.
-
HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production.J Hepatol. 2017 Apr;66(4):693-702. doi: 10.1016/j.jhep.2016.12.018. Epub 2016 Dec 24. J Hepatol. 2017. PMID: 28027970
-
The role of the NLRP3 inflammasome and the activation of IL-1β in the pathogenesis of chronic viral hepatic inflammation.Cytokine. 2018 Oct;110:389-396. doi: 10.1016/j.cyto.2018.04.032. Epub 2018 May 24. Cytokine. 2018. PMID: 29803661
-
The role of inflammasome in chronic viral hepatitis.Front Cell Infect Microbiol. 2024 May 16;14:1382029. doi: 10.3389/fcimb.2024.1382029. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 38817443 Free PMC article. Review.
-
The Role of NLRP3 Inflammasome Activation Pathway of Hepatic Macrophages in Liver Ischemia-Reperfusion Injury.Front Immunol. 2022 Jun 10;13:905423. doi: 10.3389/fimmu.2022.905423. eCollection 2022. Front Immunol. 2022. PMID: 35757691 Free PMC article. Review.
Cited by
-
HCV RNA Activates APCs via TLR7/TLR8 While Virus Selectively Stimulates Macrophages Without Inducing Antiviral Responses.Sci Rep. 2016 Jul 7;6:29447. doi: 10.1038/srep29447. Sci Rep. 2016. PMID: 27385120 Free PMC article.
-
Activation of autophagy and nucleotide-binding domain leucine-rich repeat-containing-like receptor family, pyrin domain-containing 3 inflammasome during Leishmania infantum-associated glomerulonephritis.Am J Pathol. 2015 Aug;185(8):2105-17. doi: 10.1016/j.ajpath.2015.04.017. Epub 2015 Jun 13. Am J Pathol. 2015. PMID: 26079813 Free PMC article.
-
Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities.Cell Mol Immunol. 2021 Jan;18(1):45-56. doi: 10.1038/s41423-020-00558-8. Epub 2020 Oct 12. Cell Mol Immunol. 2021. PMID: 33041338 Free PMC article. Review.
-
Hepatitis C Virus Manipulates Humans as its Favorite Host for a Long-Term Relationship.Hepatology. 2019 Feb;69(2):889-900. doi: 10.1002/hep.30214. Hepatology. 2019. PMID: 30102776 Free PMC article. Review.
-
Response of Human Liver Tissue to Innate Immune Stimuli.Front Immunol. 2022 Mar 9;13:811551. doi: 10.3389/fimmu.2022.811551. eCollection 2022. Front Immunol. 2022. PMID: 35355993 Free PMC article.
References
-
- Shepard CW, Finelli L, Alter MJ (2005) Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 5: 558–567. - PubMed
-
- Tang H, Grisé H (2009) Cellular and molecular biology of HCV infection and hepatitis. Clin Sci (Lond) 117: 49–65. - PubMed
-
- Guidotti LG, Chisari FV (2006) Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol 1: 23–61. - PubMed
-
- Dinarello CA (1984) Interleukin-1 and the pathogenesis of the acute-phase response. N Engl J Med 311: 1413–1418. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
