

High-resolution mapping of h1 linker histone variants in embryonic stem cells
- PMID: 23633960
- PMCID: PMC3636266
- DOI: 10.1371/journal.pgen.1003417
High-resolution mapping of h1 linker histone variants in embryonic stem cells
Abstract
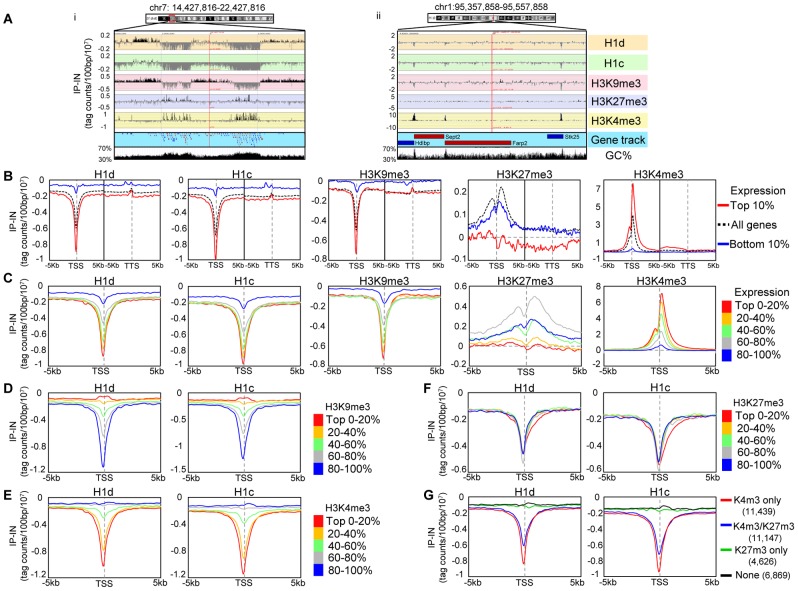
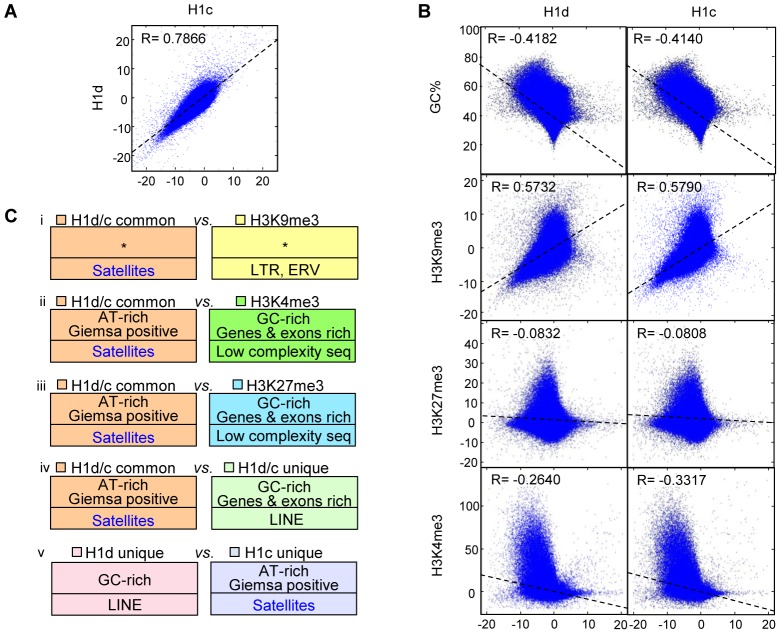
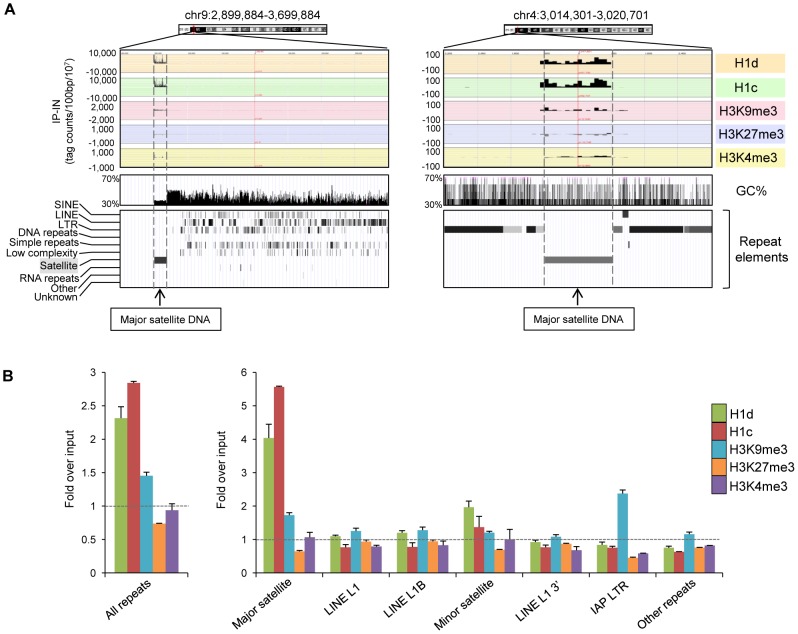
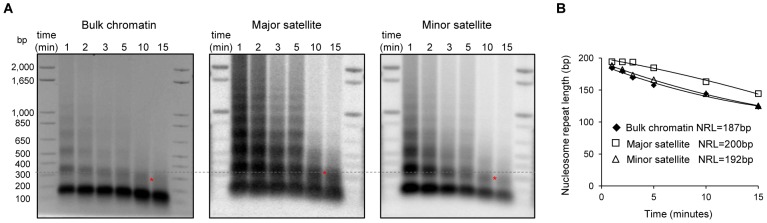
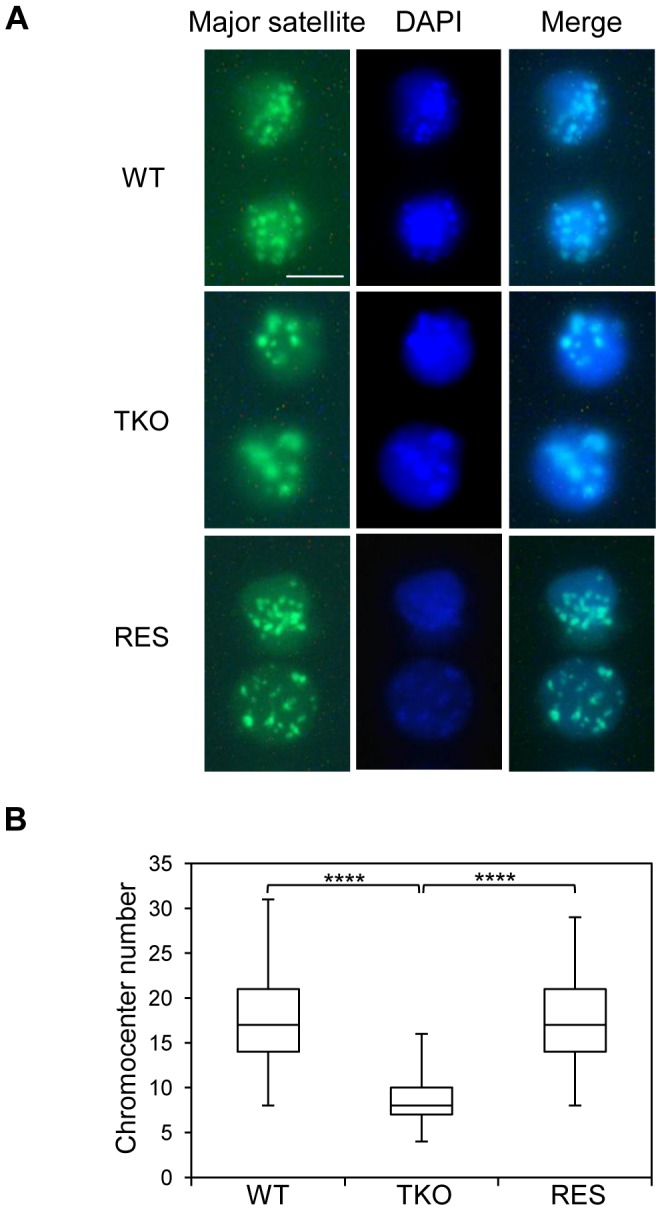
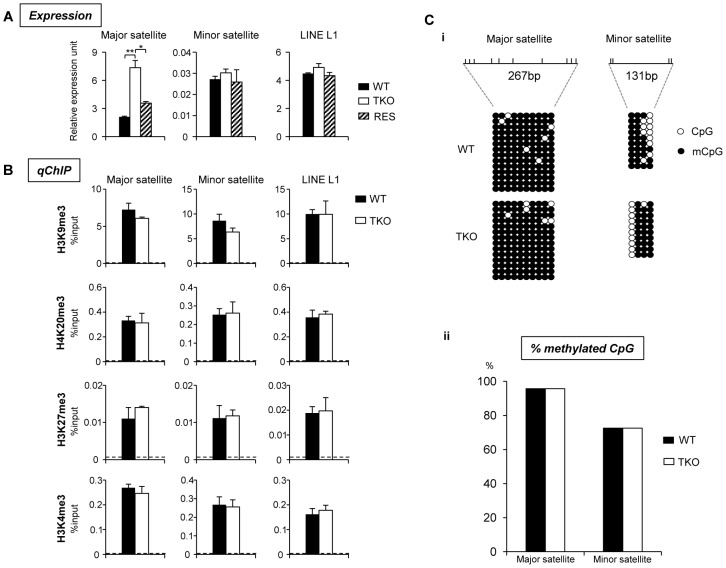
H1 linker histones facilitate higher-order chromatin folding and are essential for mammalian development. To achieve high-resolution mapping of H1 variants H1d and H1c in embryonic stem cells (ESCs), we have established a knock-in system and shown that the N-terminally tagged H1 proteins are functionally interchangeable to their endogenous counterparts in vivo. H1d and H1c are depleted from GC- and gene-rich regions and active promoters, inversely correlated with H3K4me3, but positively correlated with H3K9me3 and associated with characteristic sequence features. Surprisingly, both H1d and H1c are significantly enriched at major satellites, which display increased nucleosome spacing compared with bulk chromatin. While also depleted at active promoters and enriched at major satellites, overexpressed H1(0) displays differential binding patterns in specific repetitive sequences compared with H1d and H1c. Depletion of H1c, H1d, and H1e causes pericentric chromocenter clustering and de-repression of major satellites. These results integrate the localization of an understudied type of chromatin proteins, namely the H1 variants, into the epigenome map of mouse ESCs, and we identify significant changes at pericentric heterochromatin upon depletion of this epigenetic mark.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Wolffe AP (1998) Chromatin: Structure and Function. San Diego, CA: Academic Press.
-
- van Holde KE (1989) Chromatin: New York: Springer-Verlag.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
