

Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth
- PMID: 23635774
- PMCID: PMC3668845
- DOI: 10.1172/JCI63623
Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth
Abstract
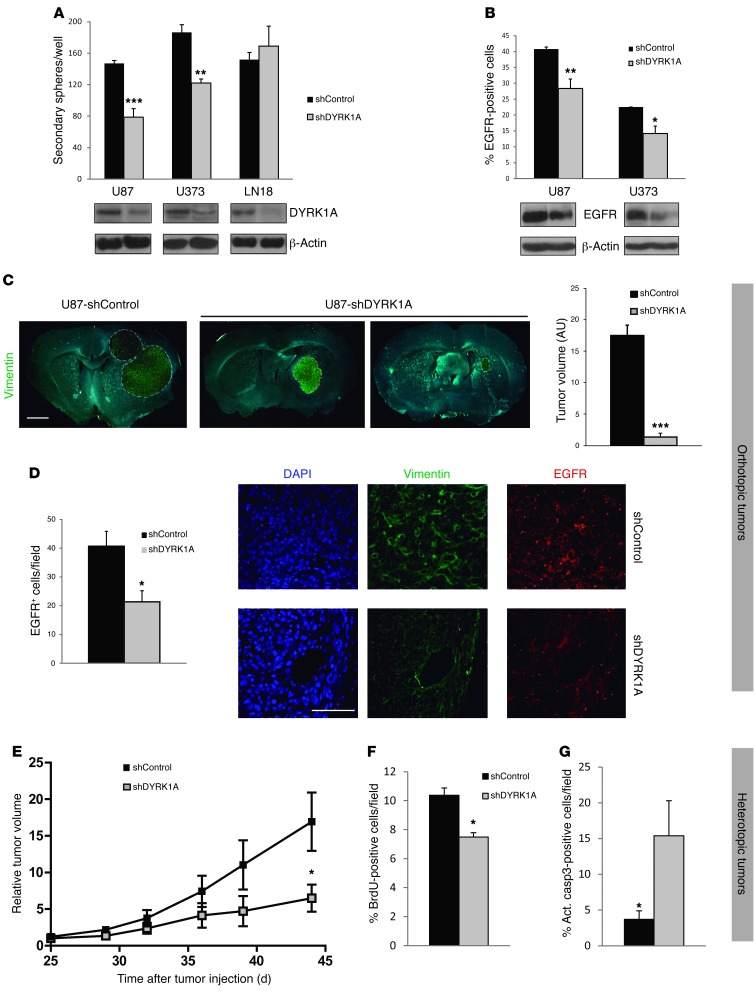
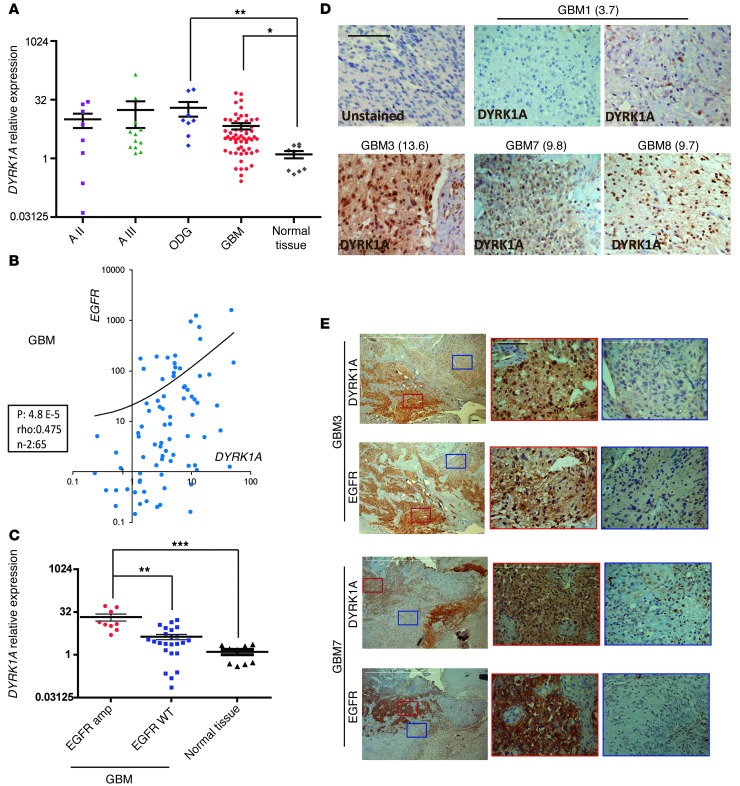
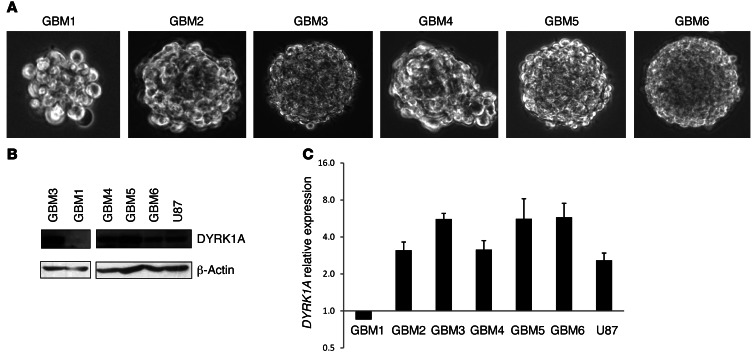
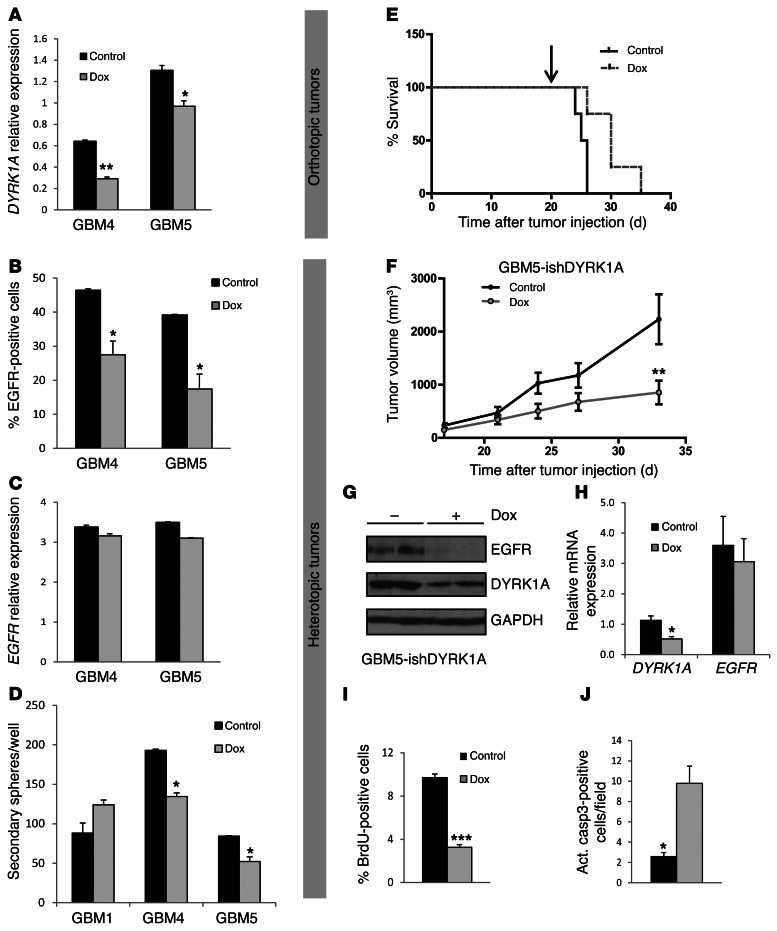
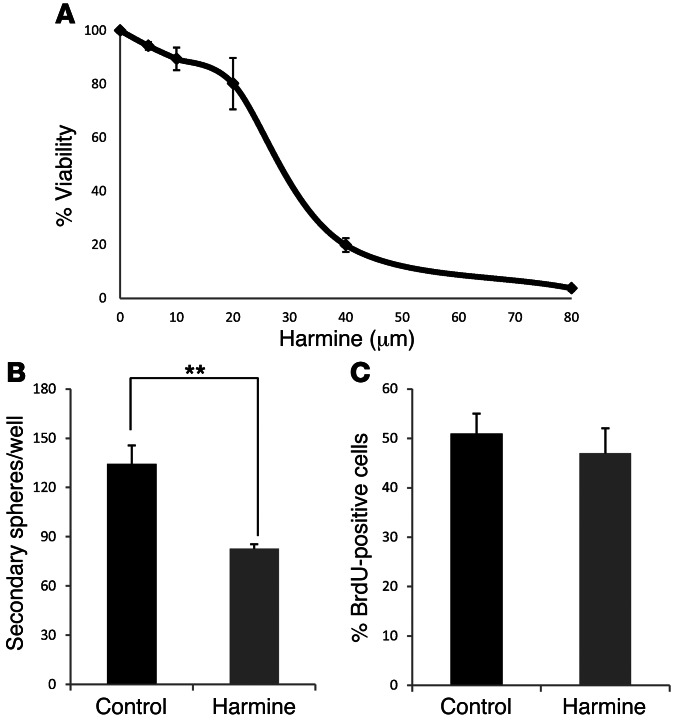
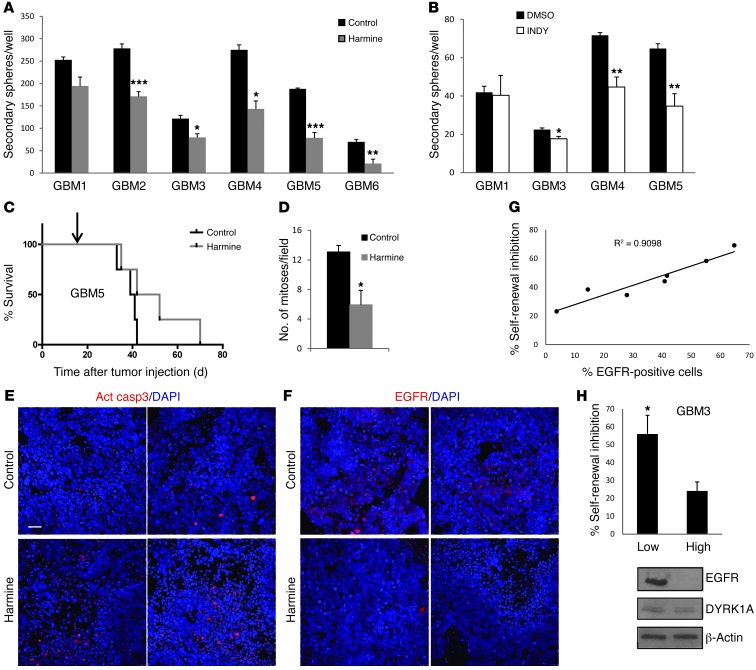
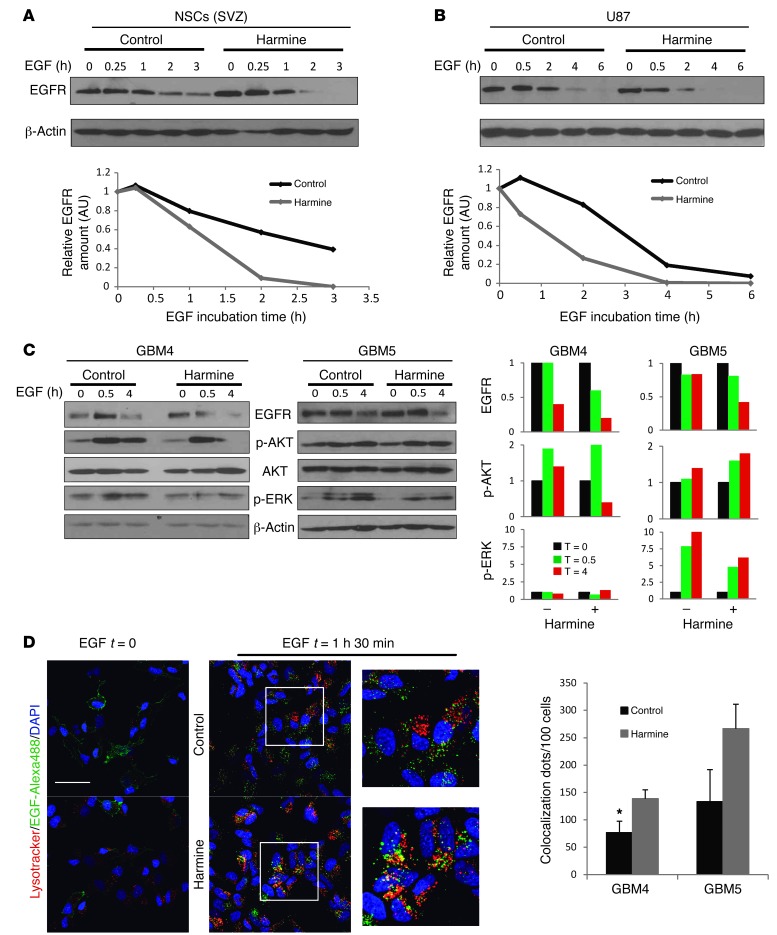
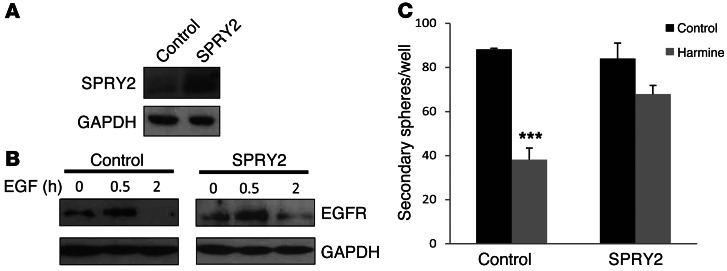
Glioblastomas (GBMs) are very aggressive tumors that are resistant to conventional chemo- and radiotherapy. New molecular therapeutic strategies are required to effectively eliminate the subpopulation of GBM tumor-initiating cells that are responsible for relapse. Since EGFR is altered in 50% of GBMs, it represents one of the most promising targets; however, EGFR kinase inhibitors have produced poor results in clinical assays, with no clear explanation for the observed resistance. We uncovered a fundamental role for the dual-specificity tyrosine phosphorylation-regulated kinase, DYRK1A, in regulating EGFR in GBMs. We found that DYRK1A was highly expressed in these tumors and that its expression was correlated with that of EGFR. Moreover, DYRK1A inhibition promoted EGFR degradation in primary GBM cell lines and neural progenitor cells, sharply reducing the self-renewal capacity of normal and tumorigenic cells. Most importantly, our data suggest that a subset of GBMs depends on high surface EGFR levels, as DYRK1A inhibition compromised their survival and produced a profound decrease in tumor burden. We propose that the recovery of EGFR stability is a key oncogenic event in a large proportion of gliomas and that pharmacological inhibition of DYRK1A could represent a promising therapeutic intervention for EGFR-dependent GBMs.
Figures
Similar articles
-
DYRK1A activates NFATC1 to increase glioblastoma migration.Cancer Med. 2021 Sep;10(18):6416-6427. doi: 10.1002/cam4.4159. Epub 2021 Jul 26. Cancer Med. 2021. PMID: 34309232 Free PMC article.
-
Dual mTORC1/2 blockade inhibits glioblastoma brain tumor initiating cells in vitro and in vivo and synergizes with temozolomide to increase orthotopic xenograft survival.Clin Cancer Res. 2014 Nov 15;20(22):5756-67. doi: 10.1158/1078-0432.CCR-13-3389. Epub 2014 Oct 14. Clin Cancer Res. 2014. PMID: 25316808
-
Evaluation of tyrosine kinase inhibitor combinations for glioblastoma therapy.PLoS One. 2012;7(10):e44372. doi: 10.1371/journal.pone.0044372. Epub 2012 Oct 2. PLoS One. 2012. PMID: 23056179 Free PMC article.
-
DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications.Pharmacol Ther. 2015 Jul;151:87-98. doi: 10.1016/j.pharmthera.2015.03.004. Epub 2015 Mar 17. Pharmacol Ther. 2015. PMID: 25795597 Review.
-
Targeting ErbB receptors in high-grade glioma.Curr Pharm Des. 2011;17(23):2468-87. doi: 10.2174/138161211797249233. Curr Pharm Des. 2011. PMID: 21827413 Review.
Cited by
-
[b]-Annulated Halogen-Substituted Indoles as Potential DYRK1A Inhibitors.Molecules. 2019 Nov 13;24(22):4090. doi: 10.3390/molecules24224090. Molecules. 2019. PMID: 31766108 Free PMC article.
-
Harmine, a dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) inhibitor induces caspase-mediated apoptosis in neuroblastoma.Cancer Cell Int. 2018 Jun 7;18:82. doi: 10.1186/s12935-018-0574-3. eCollection 2018. Cancer Cell Int. 2018. PMID: 29977157 Free PMC article.
-
Midkine signaling maintains the self-renewal and tumorigenic capacity of glioma initiating cells.Theranostics. 2020 Apr 6;10(11):5120-5136. doi: 10.7150/thno.41450. eCollection 2020. Theranostics. 2020. PMID: 32308772 Free PMC article.
-
Insights into the Clinical, Biological and Therapeutic Impact of Copy Number Alteration in Cancer.Int J Mol Sci. 2024 Jun 21;25(13):6815. doi: 10.3390/ijms25136815. Int J Mol Sci. 2024. PMID: 38999925 Free PMC article. Review.
-
A leak pathway for luminal protons in endosomes drives oncogenic signalling in glioblastoma.Nat Commun. 2015 Feb 9;6:6289. doi: 10.1038/ncomms7289. Nat Commun. 2015. PMID: 25662504 Free PMC article.
References
-
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon, France: International Agency for Research on Cancer; 2007. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
