

Thinking laterally about neurodegenerative proteinopathies
- PMID: 23635781
- PMCID: PMC3635732
- DOI: 10.1172/JCI66029
Thinking laterally about neurodegenerative proteinopathies
Abstract
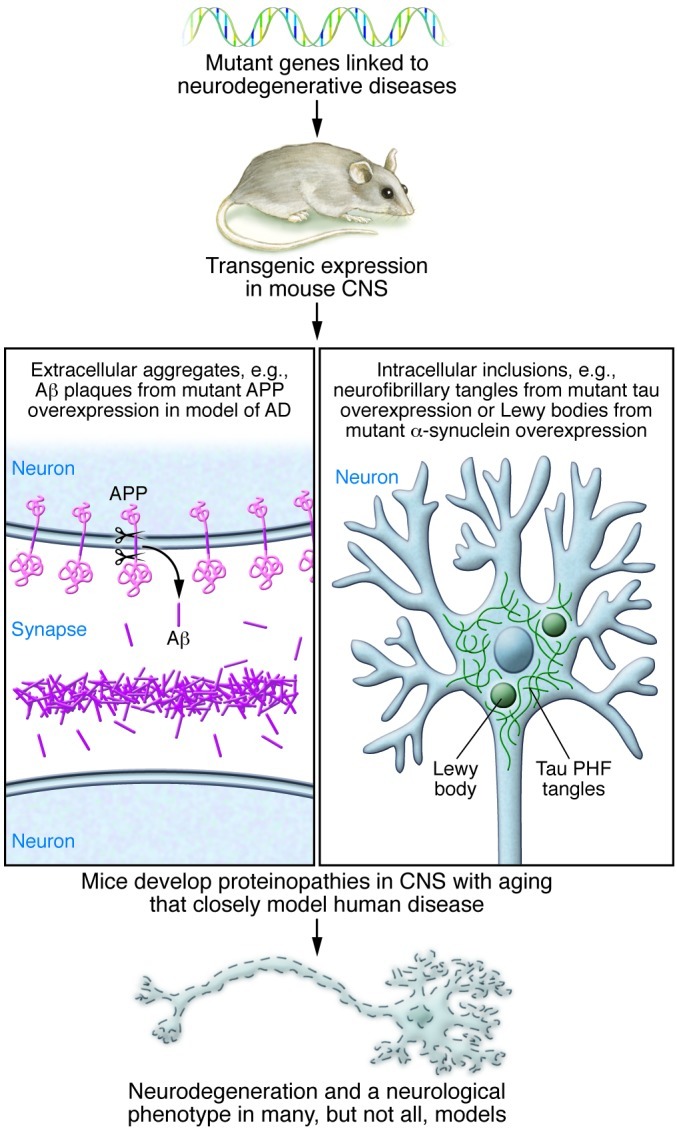
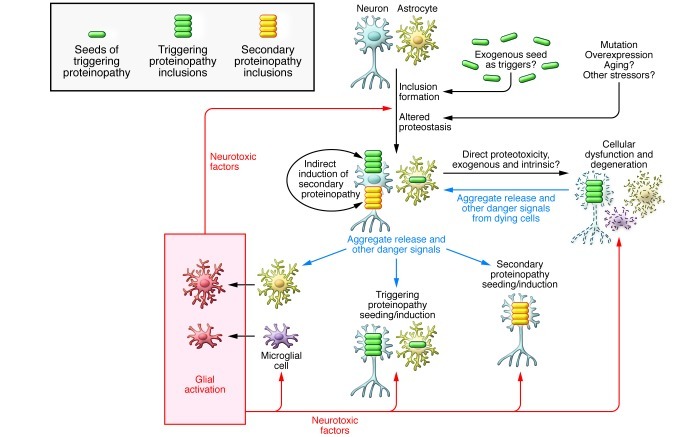
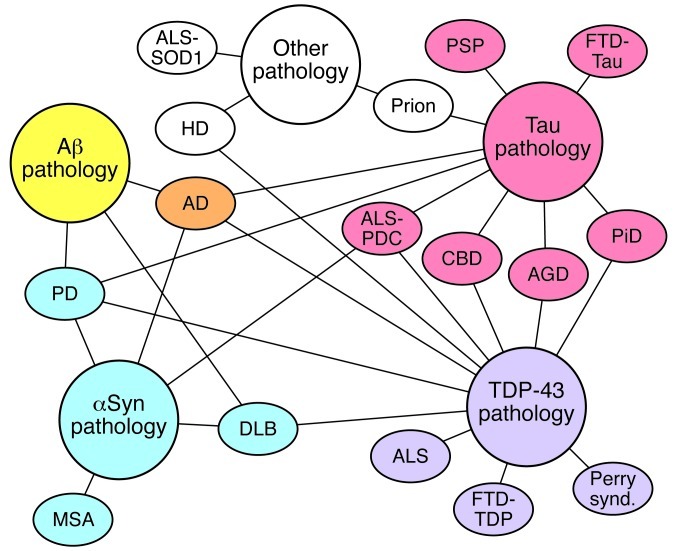
Many neurodegenerative disorders, including Alzheimer's disease, Parkinson's disease, Huntington's disease, and frontotemporal dementia, are proteinopathies that are associated with the aggregation and accumulation of misfolded proteins. While remarkable progress has been made in understanding the triggers of these conditions, several challenges have hampered the translation of preclinical therapies targeting pathways downstream of the initiating proteinopathies. Clinical trials in symptomatic patients using therapies directed toward initiating trigger events have met with little success, prompting concerns that such therapeutics may be of limited efficacy when used in advanced stages of the disease rather than as prophylactics. Herein, we discuss gaps in our understanding of the pathological processes downstream of the trigger and potential strategies to identify common features of the downstream degenerative cascade in multiple CNS proteinopathies, which could potentially lead to the development of common therapeutic targets for multiple disorders.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
