

Defective K-Ras oncoproteins overcome impaired effector activation to initiate leukemia in vivo
- PMID: 23637129
- PMCID: PMC3682340
- DOI: 10.1182/blood-2012-05-432252
Defective K-Ras oncoproteins overcome impaired effector activation to initiate leukemia in vivo
Abstract
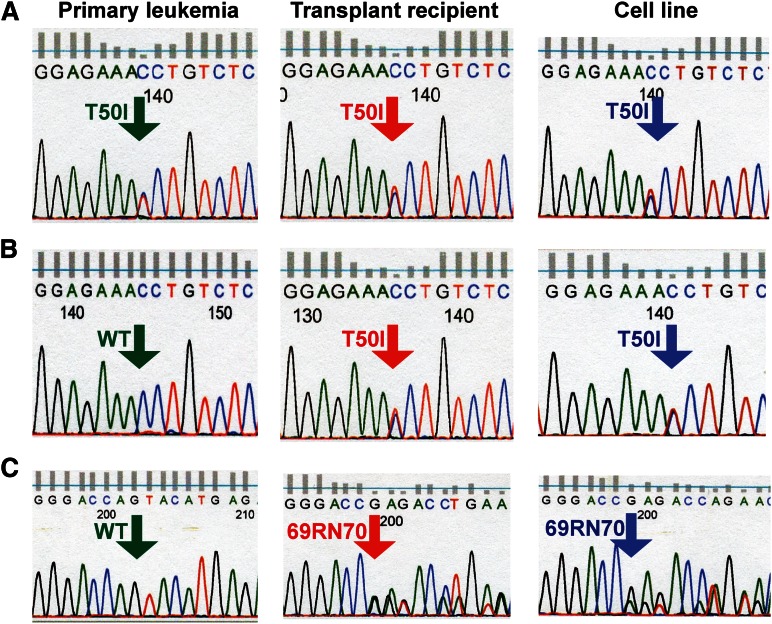
Reversing the aberrant biochemical output of oncogenic Ras proteins is one of the great challenges in cancer therapeutics; however, it is uncertain which Ras effectors are required for tumor initiation and maintenance. To address this question, we expressed oncogenic K-Ras(D12) proteins with "second site" amino acid substitutions that impair PI3 kinase/Akt or Raf/MEK/ERK activation in bone marrow cells and transplanted them into recipient mice. In spite of attenuated signaling properties, defective K-Ras oncoproteins initiated aggressive clonal T-lineage acute lymphoblastic leukemia (T-ALL). Murine T-ALLs expressing second site mutant proteins restored full oncogenic Ras activity through diverse mechanisms, which included acquiring novel somatic third site Kras(D12) mutations and silencing PTEN. T-ALL cell lines lacking PTEN had elevated levels of phosphorylated Akt, a gene expression pattern similar to human early T-cell precursor ALL, and were resistant to the potent and selective MEK inhibitor PD0325901. Our data, which demonstrate strong selective pressure to overcome the defective activation of PI3 kinase/Akt and Raf/MEK/ERK, implicate both Ras effector pathways as drivers of aberrant growth in T-ALL and further suggest that leukemia cells will deploy multiple mechanisms to develop resistance to targeted inhibitors in vivo.
Figures





References
-
- Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294(5545):1299–1304. - PubMed
-
- Donovan S, Shannon KM, Bollag G. GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta. 2002;1602(1):23–45. - PubMed
-
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
