

Death receptor-ligand systems in cancer, cell death, and inflammation
- PMID: 23637280
- PMCID: PMC3632055
- DOI: 10.1101/cshperspect.a008698
Death receptor-ligand systems in cancer, cell death, and inflammation
Abstract
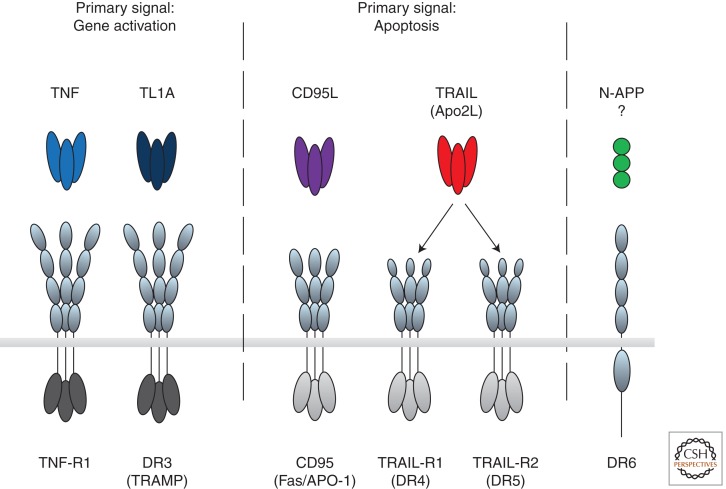
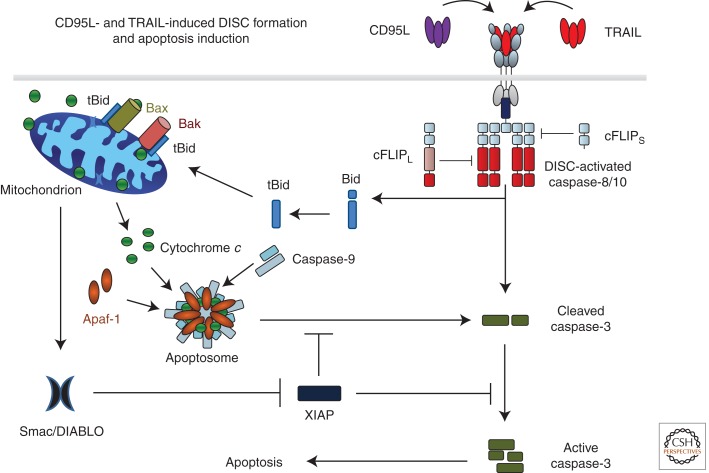
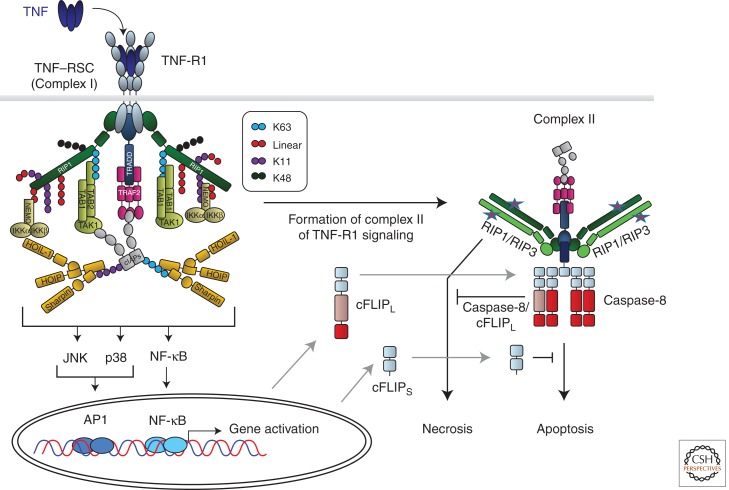
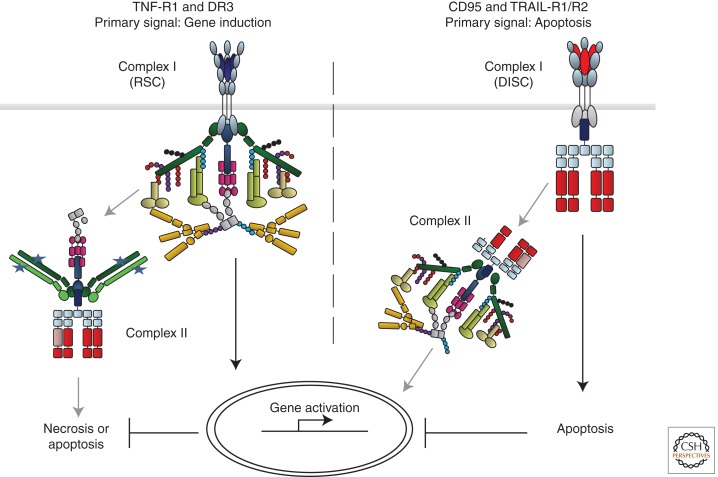
The discovery of tumor necrosis factor (TNF) marked the beginning of one of the most fascinating journeys in modern biomedical research. For the moment, this journey has culminated in the development of drugs that inhibit TNF. TNF blockers have revolutionized the treatment of many chronic inflammatory diseases. Yet, the journey seems far from over. TNF is the founding member of a family of cytokines with crucial functions in cell death, inflammation, and cancer. Some of these factors, most prominently TNF, CD95L, and TRAIL, can induce cell death. The receptors that mediate this signal are therefore referred to as death receptors, even though they also activate other signals. Here I will take you on a journey into the discovery and study of death receptor-ligand systems and how this inspired new concepts in cancer therapy and our current understanding of the interplay between cell death and inflammation.
Figures
References
-
- Ackery A, Robins S, Fehlings MG 2006. Inhibition of Fas-mediated apoptosis through administration of soluble Fas receptor improves functional outcome and reduces posttraumatic axonal degeneration after acute spinal cord injury. J Neurotrauma 23: 604–616 - PubMed
-
- Alderson MR, Tough TW, Braddy S, Davis-Smith T, Roux E, Schooley K, Miller RE, Lynch DH 1994. Regulation of apoptosis and T cell activation by Fas-specific mAb. Int Immunol 6: 1799–1806 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources