

Review
doi: 10.1101/cshperspect.a010256.
Chlamydial intracellular survival strategies
Affiliations
- PMID: 23637308
- PMCID: PMC3633179
- DOI: 10.1101/cshperspect.a010256
Item in Clipboard
Review
Chlamydial intracellular survival strategies
Cold Spring Harb Perspect Med.
.
Abstract
Chlamydia trachomatis is the most common sexually transmitted bacterial pathogen and the causative agent of blinding trachoma. Although Chlamydia is protected from humoral immune responses by residing within remodeled intracellular vacuoles, it still must contend with multilayered intracellular innate immune defenses deployed by its host while scavenging for nutrients. Here we provide an overview of Chlamydia biology and highlight recent findings detailing how this vacuole-bound pathogen manipulates host-cellular functions to invade host cells and maintain a replicative niche.
Figures
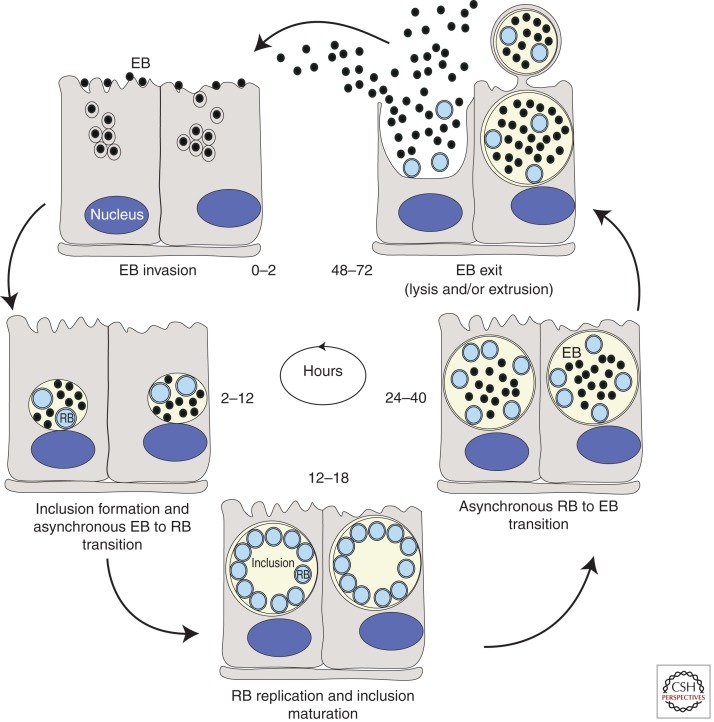
Life cycle of Chlamydia. Within the first 2 h following internalization into cells, elementary bodies (EBs) fuse to form a nascent inclusion. Between 2 and 6 h postinternalization, EBs begin to differentiate into reticulate bodies (RBs). By 12 h postinfection (hpi) RBs can be observed dividing by binary fission and by 18–24 hpi they peak in numbers. Increasing numbers of RBs differentiate back to EBs around 24 hpi and continue differentiating until lysis or release occurs ∼48–72 hpi depending on the chlamydial species (Hatch 1999).
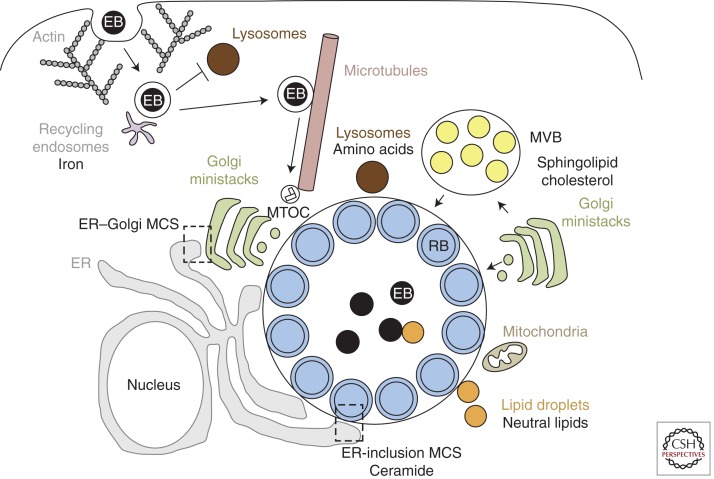
Chlamydia and host–cellular interactions. Chlamydia enters host cells by an actin-dependent mechanism that involves both host and bacterial factors. The EBs are enclosed within a membrane-bound inclusion, which becomes modified by insertion of bacterial inclusion proteins (Incs) that prevent fusion with lysosomes and promote interactions with recycling endosomes for iron acquisition. The nascent inclusion is transported along microtubules to the MTOC and EBs transition into RBs. The inclusion expands to accommodate the replicating RBs. During this time, the inclusion interacts, likely via bacterial effectors, with multiple host-cell organelles, including fragmented Golgi ministacks, the ER, lipid droplets, MVBs, and the mitochondria. Delivery of essential host lipids to the inclusion involves vesicular trafficking from Golgi ministacks and MVBs as well as nonvesicular trafficking from lipid droplets and membrane contact sites (MCS) formed between the ER and the inclusion. Although the inclusion does not fuse with lysosomes, Chlamydia may import essential amino acids derived from host-protein degradation within lysosomes.
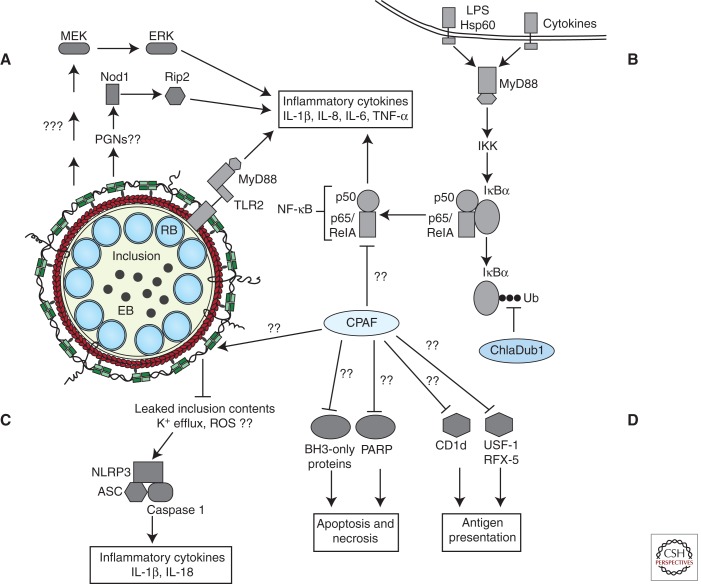
Current model of chlamydial effector interactions with host-innate immune signaling pathways. (A) TLR2 and MyD88 are trafficked to the inclusion by unknown mechanisms and promote expression of inflammatory cytokines. Nod1 and MEK-ERK MAPK signaling pathways also stimulate inflammatory cytokine gene expression. (B) Chlamydial microbial products and cytokines secreted by recruited immune cells and neighboring infected cells activate Toll-like and cytokine receptors. Receptor signaling activates NF-κB signaling and expression of proinflammatory cytokines/chemokines. Established inclusions impede NF-κB signaling by reversing IκBα ubiquitination through the activity of secreted ChlaDub1 and possibly by CPAF-mediated degradation of p65/RelA. (C) A cytoskeleton cage comprised of actin (red) and intermediate filaments (green) envelops the established inclusion. Remodeling of the cage curtails inclusion contents from activating cytosolic PRRs, possibly the NLRP3-ASC-caspase 1 inflammasome. However, unknown chlamydial agonists can activate the NLRP3-ASC inflammasome and caspase 1-dependent processing of inflammatory cytokines. (D) Additional mechanisms facilitating host survival may include the degradation of BH3-only proteins and PARP (Yu et al. 2010) and blocking death-inducing signaling pathways presumably originating from the TNF family of receptors and cytokine receptors. CPAF activity may also be deployed by Chlamydia to subvert antigen presentation.
References
-
- Balana ME, Niedergang F, Subtil A, Alcover A, Chavrier P, Dautry-Varsat A 2005. ARF6 GTPase controls bacterial invasion by actin remodelling. J Cell Sci 118: 2201–2210 - PubMed
-
- Beatty WL 2006. Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J Cell Sci 119: 350–359 - PubMed
-
- Beatty WL 2007. Lysosome repair enables host cell survival and bacterial persistence following Chlamydia trachomatis infection. Cell Microbiol 9: 2141–2152 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical