

Review
doi: 10.1101/cshperspect.a011700.
The molecular basis of β-thalassemia
Affiliations
- PMID: 23637309
- PMCID: PMC3633182
- DOI: 10.1101/cshperspect.a011700
Item in Clipboard
Review
The molecular basis of β-thalassemia
Cold Spring Harb Perspect Med.
.
Abstract
The β-thalassemias are characterized by a quantitative deficiency of β-globin chains underlaid by a striking heterogeneity of molecular defects. Although most of the molecular lesions involve the structural β gene directly, some down-regulate the gene through distal cis effects, and rare trans-acting mutations have also been identified. Most β-thalassemias are inherited in a Mendelian recessive fashion but there is a subgroup of β-thalassemia alleles that behave as dominant negatives. Unraveling the molecular basis of β-thalassemia has provided a paradigm for understanding of much of human genetics.
Figures
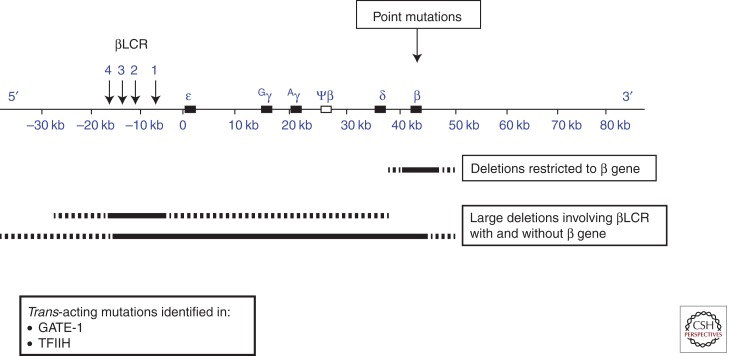
Mutations causing β-thalassemia. The upper panel depicts the β-globin gene cluster with the upstream locus control region (βLCR). The mutations can be cis-acting and include point mutations affecting the structural β gene, deletions restricted to the β gene, and large deletions involving the βLCR with or without the β gene. The dashed lines represent variations in the amount of flanking DNA removed by the deletions—detailed in Figures 3 and 4, respectively.
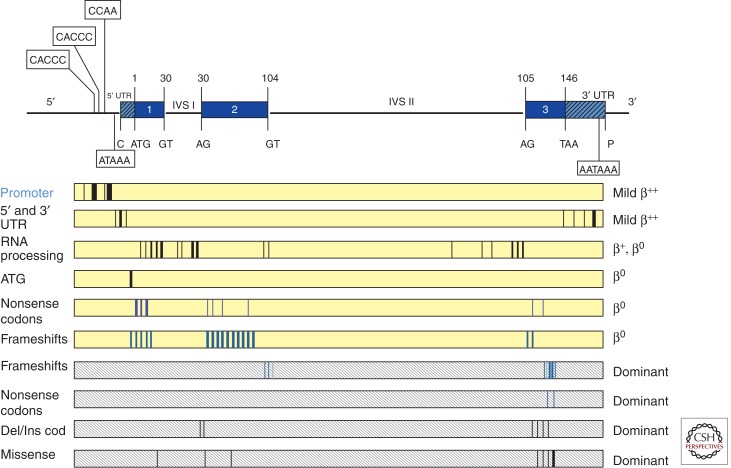
Point mutations causing β-thalassemia. The β-globin gene is depicted in the upper panel with conserved sequences in the 5′ and 3′ UTRs, and the invariant dinucleotides in exon–intron junctions of the gene, important in the control of gene expression. The boxes represent the different categories of mutants. The vertical lines within the boxes represent the sites of the different mutations. Dominantly inherited mutants are found within shaded boxes.
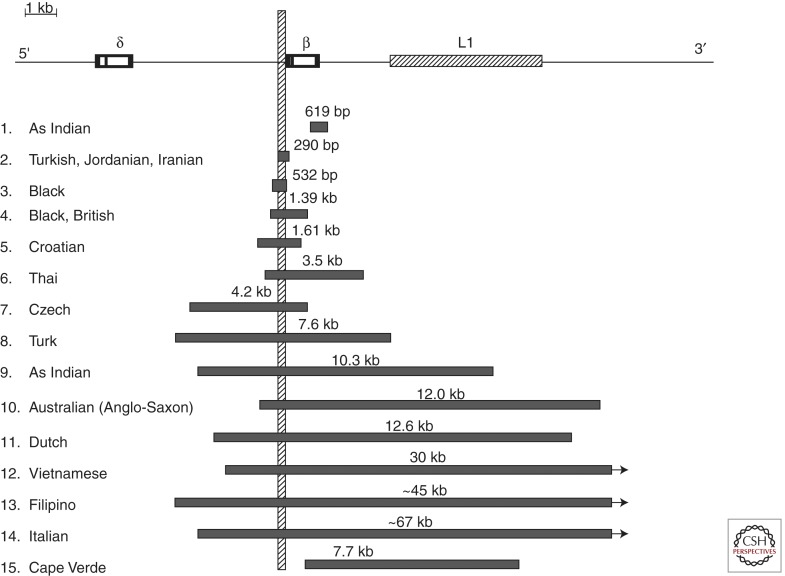
β-thalassemia deletions that are restricted to the gene. The vertical bar indicates the promoter region that is removed in common by these deletions except for the 619 bp Asian Indian and the 7.7 kb Cape Verde deletions.

Deletions causing β-thalassemia as part of (εγδβ)0-thalassemia. The deletions can be classified into two groups: group I deletions (in black) remove all or most of the cluster including the βLCR and the β-globin gene, whereas group II deletions remove all or part of βLCR leaving the β gene intact. Only about one-third of the breakpoints of these deletions have been defined; the white boxes and jagged ends indicate undefined breakpoints.
References
-
- Andersson BA, Wering ME, Luo HY, Basran RK, Steinberg MH, Smith HP, Chui DH 2007. Sickle cell disease due to compound heterozygosity for Hb S and a novel 7.7-kb β-globin gene deletion. Eur J Haematol 78: 82–85 - PubMed
-
- Arjona SN, Maldonado Eloy-Garcia J, Gu L-H, Smetanina NS, Huisman THJ 1996. The dominant β-thalassaemia in a Spanish family is due to a frameshift that introduces an extra CGG codon (=arginine) at the 5′ end of the second exon. Br J Haematol 93: 841–844 - PubMed
-
- Badens C, Mattei MG, Imbert AM, Lapoumérouliee C, Martini N, Michel G, Lena-Russo D 2002. A novel mechanism for thalassaemia intermedia. Lancet 359: 132–133 - PubMed
-
- Baiget M, Gomez Pereira C, Jue DL, Johnson MH, McGuffey JE, Moo-Penn WF 1986. A case of hemoglobin Indianapolis [β112(G14) Cys → Arg] in an individual from Cordoba, Spain. Hemoglobin 10: 483–494 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical