

A mechanistic paradigm for broad-spectrum antivirals that target virus-cell fusion
- PMID: 23637597
- PMCID: PMC3630091
- DOI: 10.1371/journal.ppat.1003297
A mechanistic paradigm for broad-spectrum antivirals that target virus-cell fusion
Abstract
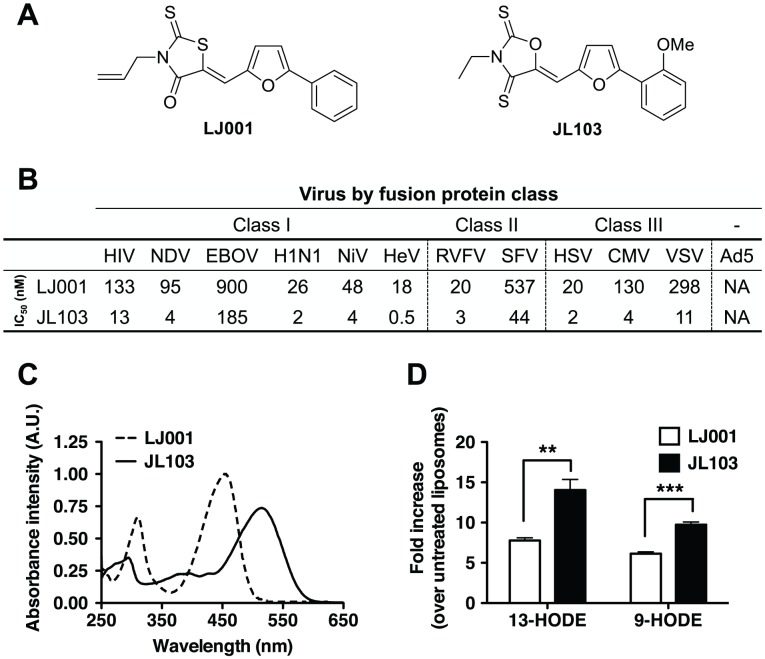
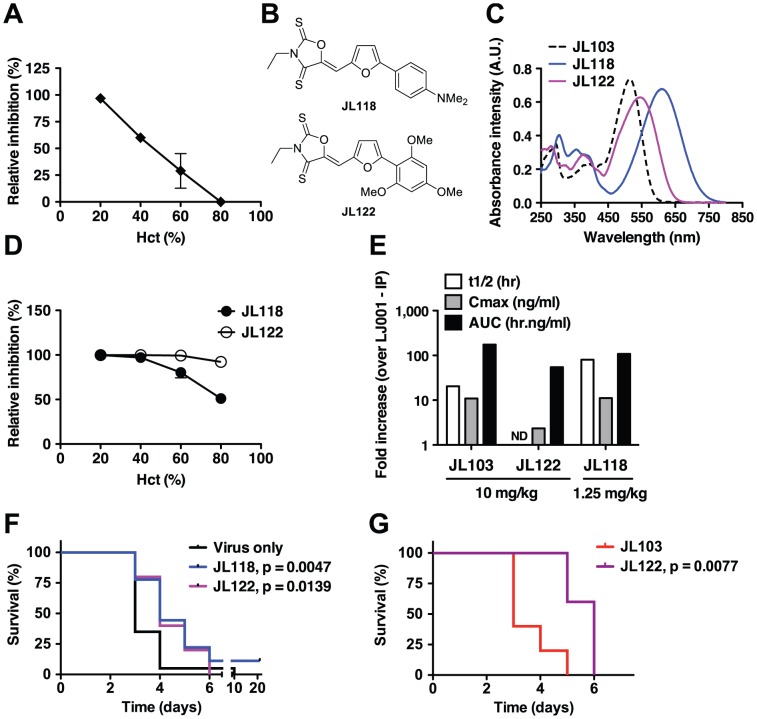
LJ001 is a lipophilic thiazolidine derivative that inhibits the entry of numerous enveloped viruses at non-cytotoxic concentrations (IC50 ≤ 0.5 µM), and was posited to exploit the physiological difference between static viral membranes and biogenic cellular membranes. We now report on the molecular mechanism that results in LJ001's specific inhibition of virus-cell fusion. The antiviral activity of LJ001 was light-dependent, required the presence of molecular oxygen, and was reversed by singlet oxygen ((1)O2) quenchers, qualifying LJ001 as a type II photosensitizer. Unsaturated phospholipids were the main target modified by LJ001-generated (1)O2. Hydroxylated fatty acid species were detected in model and viral membranes treated with LJ001, but not its inactive molecular analog, LJ025. (1)O2-mediated allylic hydroxylation of unsaturated phospholipids leads to a trans-isomerization of the double bond and concurrent formation of a hydroxyl group in the middle of the hydrophobic lipid bilayer. LJ001-induced (1)O2-mediated lipid oxidation negatively impacts on the biophysical properties of viral membranes (membrane curvature and fluidity) critical for productive virus-cell membrane fusion. LJ001 did not mediate any apparent damage on biogenic cellular membranes, likely due to multiple endogenous cytoprotection mechanisms against phospholipid hydroperoxides. Based on our understanding of LJ001's mechanism of action, we designed a new class of membrane-intercalating photosensitizers to overcome LJ001's limitations for use as an in vivo antiviral agent. Structure activity relationship (SAR) studies led to a novel class of compounds (oxazolidine-2,4-dithiones) with (1) 100-fold improved in vitro potency (IC50<10 nM), (2) red-shifted absorption spectra (for better tissue penetration), (3) increased quantum yield (efficiency of (1)O2 generation), and (4) 10-100-fold improved bioavailability. Candidate compounds in our new series moderately but significantly (p≤0.01) delayed the time to death in a murine lethal challenge model of Rift Valley Fever Virus (RVFV). The viral membrane may be a viable target for broad-spectrum antivirals that target virus-cell fusion.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
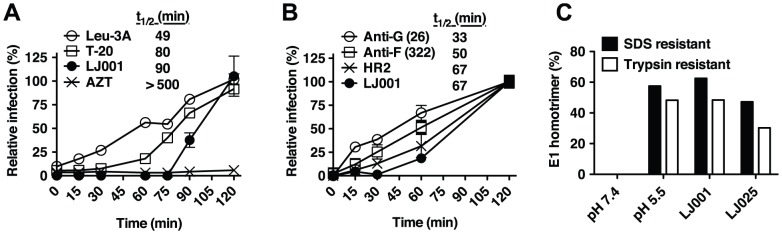
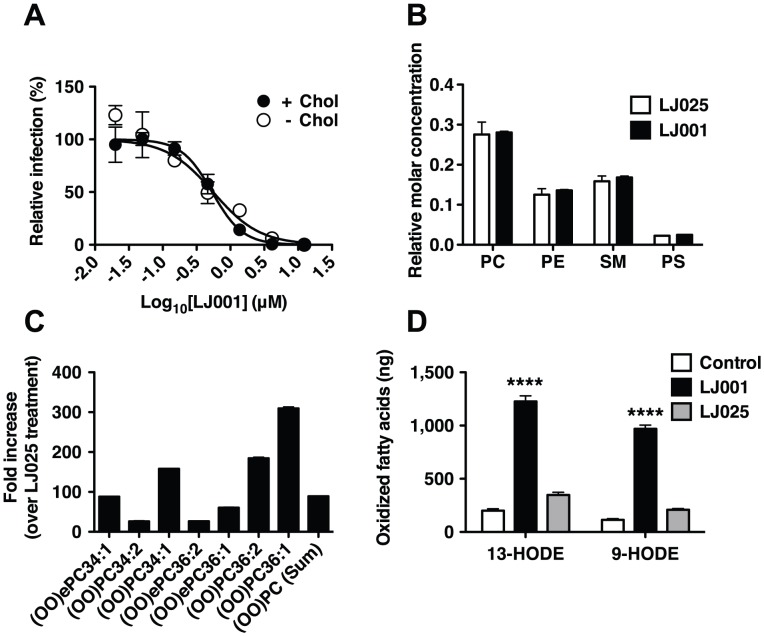
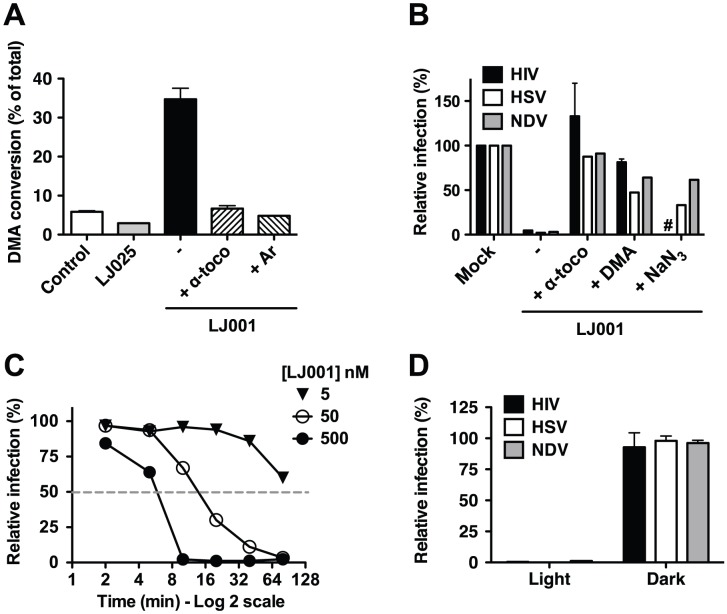
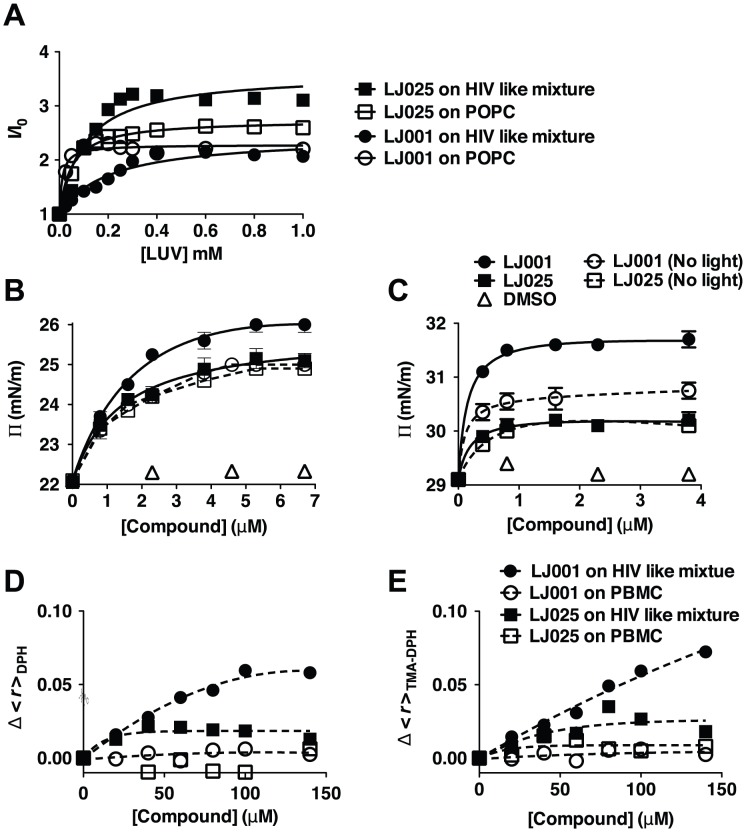
References
-
- Kesel AJ (2011) Broad-spectrum antiviral activity including human immunodeficiency and hepatitis C viruses mediated by a novel retinoid thiosemicarbazone derivative. Eur J Med Chem 46: 1656–1664. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
