

The genome organization of Thermotoga maritima reflects its lifestyle
- PMID: 23637642
- PMCID: PMC3636130
- DOI: 10.1371/journal.pgen.1003485
The genome organization of Thermotoga maritima reflects its lifestyle
Abstract
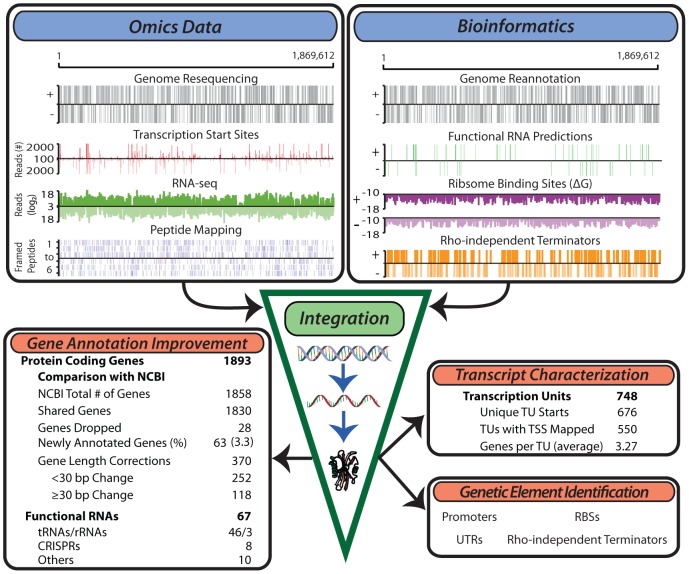
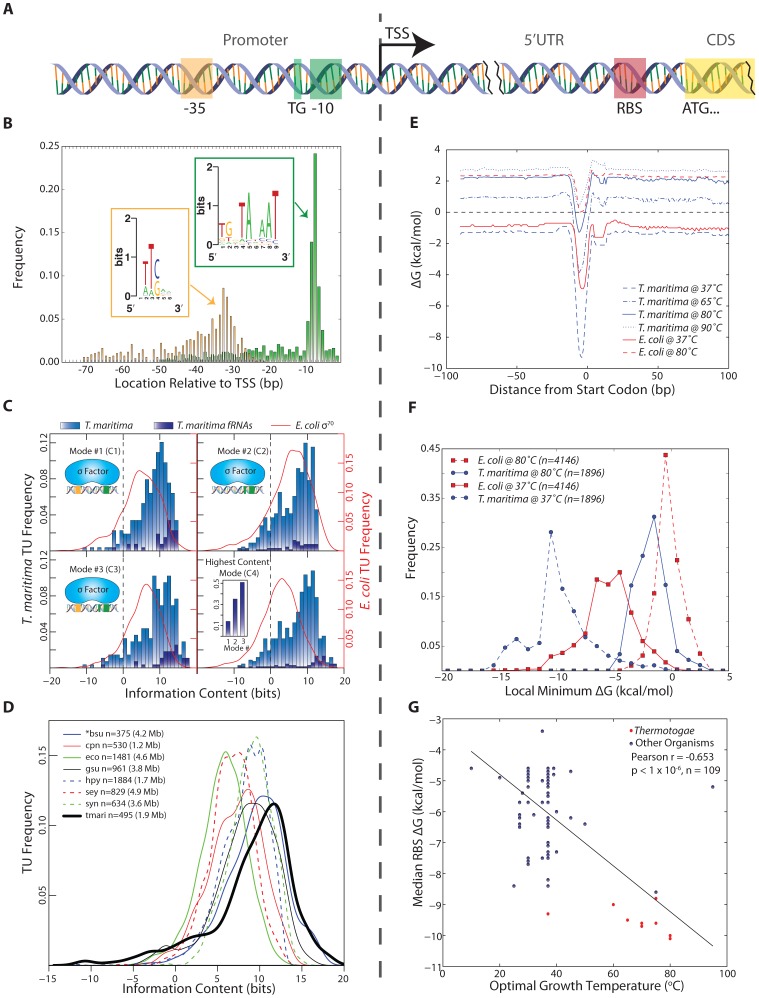
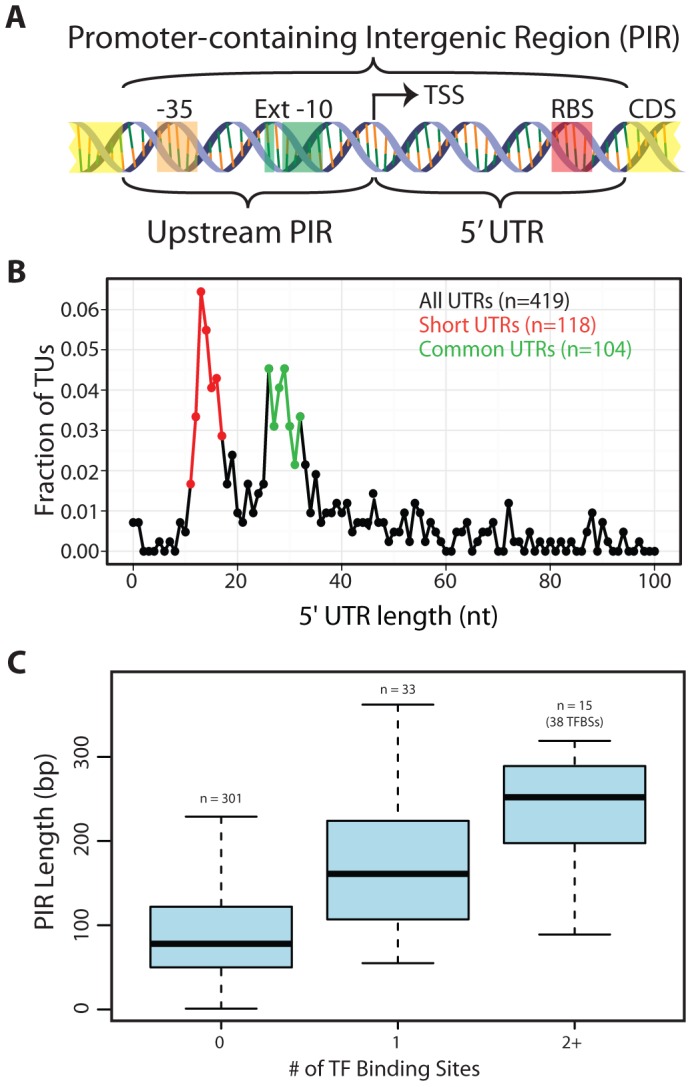
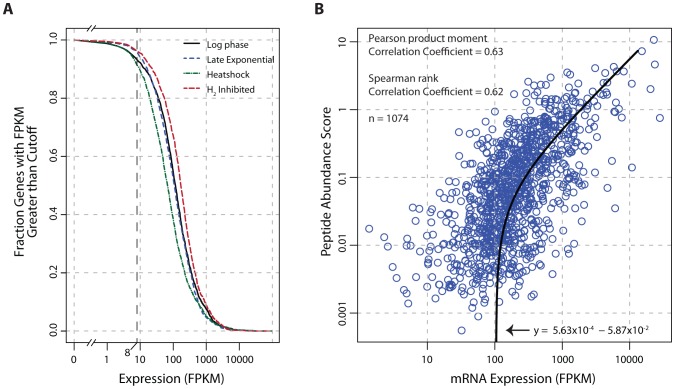
The generation of genome-scale data is becoming more routine, yet the subsequent analysis of omics data remains a significant challenge. Here, an approach that integrates multiple omics datasets with bioinformatics tools was developed that produces a detailed annotation of several microbial genomic features. This methodology was used to characterize the genome of Thermotoga maritima--a phylogenetically deep-branching, hyperthermophilic bacterium. Experimental data were generated for whole-genome resequencing, transcription start site (TSS) determination, transcriptome profiling, and proteome profiling. These datasets, analyzed in combination with bioinformatics tools, served as a basis for the improvement of gene annotation, the elucidation of transcription units (TUs), the identification of putative non-coding RNAs (ncRNAs), and the determination of promoters and ribosome binding sites. This revealed many distinctive properties of the T. maritima genome organization relative to other bacteria. This genome has a high number of genes per TU (3.3), a paucity of putative ncRNAs (12), and few TUs with multiple TSSs (3.7%). Quantitative analysis of promoters and ribosome binding sites showed increased sequence conservation relative to other bacteria. The 5'UTRs follow an atypical bimodal length distribution comprised of "Short" 5'UTRs (11-17 nt) and "Common" 5'UTRs (26-32 nt). Transcriptional regulation is limited by a lack of intergenic space for the majority of TUs. Lastly, a high fraction of annotated genes are expressed independent of growth state and a linear correlation of mRNA/protein is observed (Pearson r = 0.63, p<2.2 × 10(-16) t-test). These distinctive properties are hypothesized to be a reflection of this organism's hyperthermophilic lifestyle and could yield novel insights into the evolutionary trajectory of microbial life on earth.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Kitano H (2002) Systems biology: a brief overview. Science 295: 1662–1664. - PubMed
-
- Reed JL, Famili I, Thiele I, Palsson BO (2006) Towards multidimensional genome annotation. Nat Rev Genet 7: 130–141. - PubMed
-
- Overbeek R, Bartels D, Vonstein V, Meyer F (2007) Annotation of bacterial and archaeal genomes: improving accuracy and consistency. Chem Rev 107: 3431–3447. - PubMed
-
- Guell M, van Noort V, Yus E, Chen WH, Leigh-Bell J, et al. (2009) Transcriptome complexity in a genome-reduced bacterium. Science 326: 1268–1271. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
