

The genome sequence of Lone Star virus, a highly divergent bunyavirus found in the Amblyomma americanum tick
- PMID: 23637969
- PMCID: PMC3639253
- DOI: 10.1371/journal.pone.0062083
The genome sequence of Lone Star virus, a highly divergent bunyavirus found in the Amblyomma americanum tick
Abstract
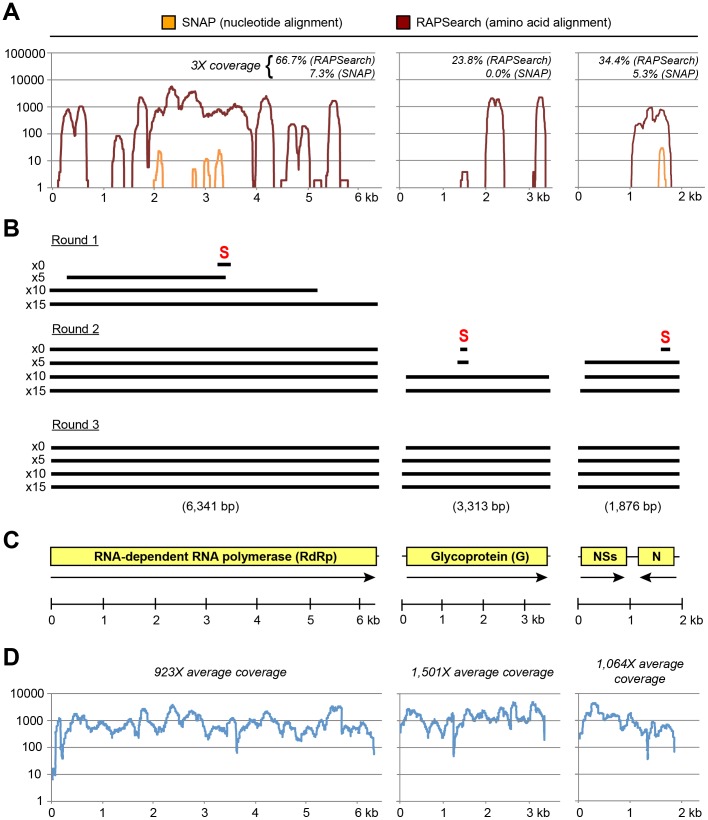
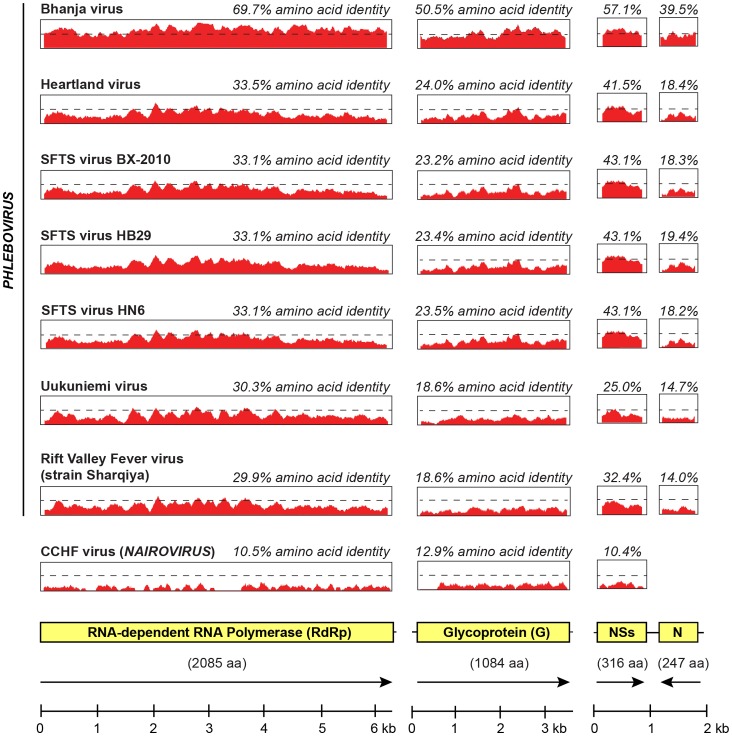
Viruses in the family Bunyaviridae infect a wide range of plant, insect, and animal hosts. Tick-borne bunyaviruses in the Phlebovirus genus, including Severe Fever with Thrombocytopenia Syndrome virus (SFTSV) in China, Heartland virus (HRTV) in the United States, and Bhanja virus in Eurasia and Africa have been associated with acute febrile illness in humans. Here we sought to characterize the growth characteristics and genome of Lone Star virus (LSV), an unclassified bunyavirus originally isolated from the lone star tick Amblyomma americanum. LSV was able to infect both human (HeLa) and monkey (Vero) cells. Cytopathic effects were seen within 72 h in both cell lines; vacuolization was observed in infected Vero, but not HeLa, cells. Viral culture supernatants were examined by unbiased deep sequencing and analysis using an in-house developed rapid computational pipeline for viral discovery, which definitively identified LSV as a phlebovirus. De novo assembly of the full genome revealed that LSV is highly divergent, sharing <61% overall amino acid identity with any other bunyavirus. Despite this sequence diversity, LSV was found by phylogenetic analysis to be part of a well-supported clade that includes members of the Bhanja group viruses, which are most closely related to SFSTV/HRTV. The genome sequencing of LSV is a critical first step in developing diagnostic tools to determine the risk of arbovirus transmission by A. americanum, a tick of growing importance given its expanding geographic range and competence as a disease vector. This study also underscores the power of deep sequencing analysis in rapidly identifying and sequencing the genomes of viruses of potential clinical and public health significance.
Conflict of interest statement
Figures


References
-
- Walter CT, Barr JN (2011) Recent advances in the molecular and cellular biology of bunyaviruses. J Gen Virol 92: 2467–2484. - PubMed
-
- Ergonul O (2012) Crimean-Congo hemorrhagic fever virus: new outbreaks, new discoveries. Curr Opin Virol 2: 215–220. - PubMed
-
- Guu TS, Zheng W, Tao YJ (2012) Bunyavirus: structure and replication. Adv Exp Med Biol 726: 245–266. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
