

Neutron encoded labeling for peptide identification
- PMID: 23638792
- PMCID: PMC3827945
- DOI: 10.1021/ac400476w
Neutron encoded labeling for peptide identification
Abstract
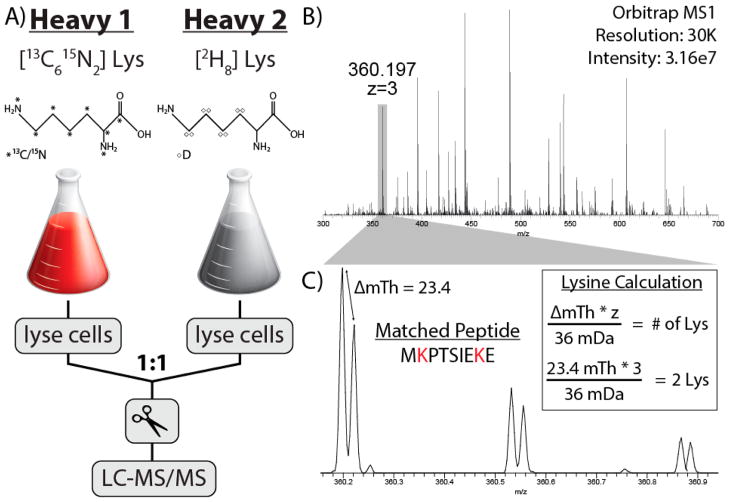
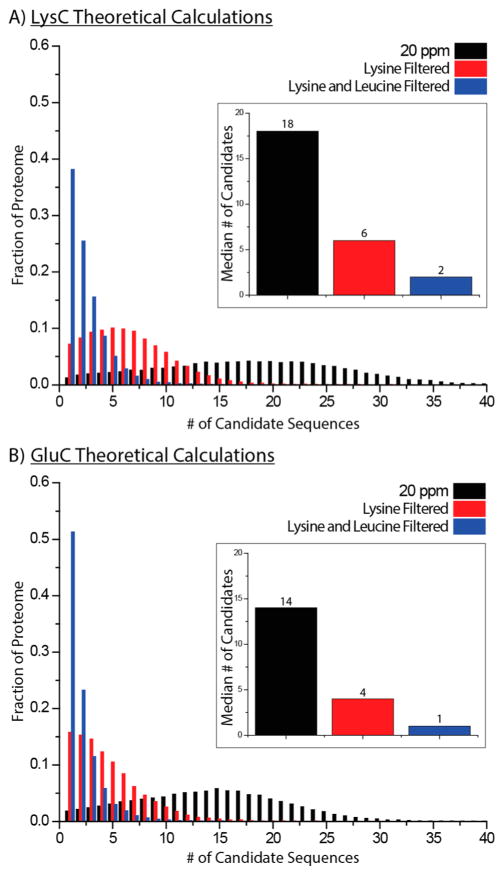
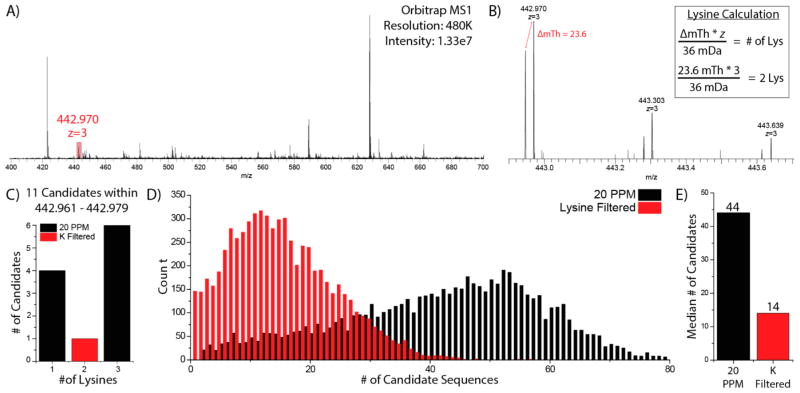
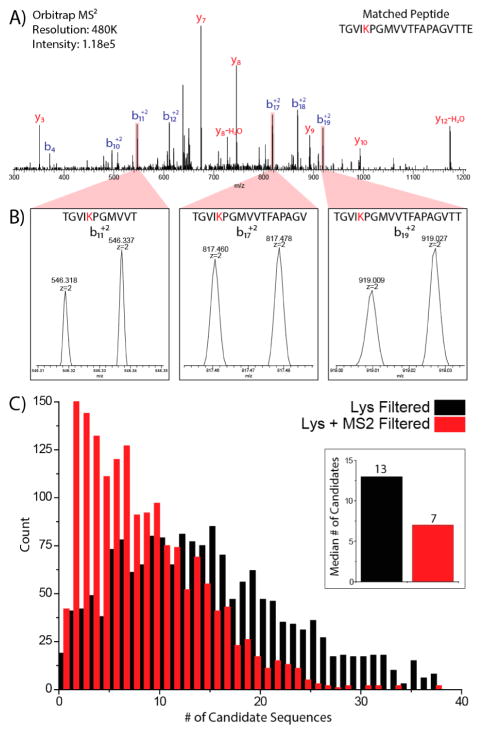
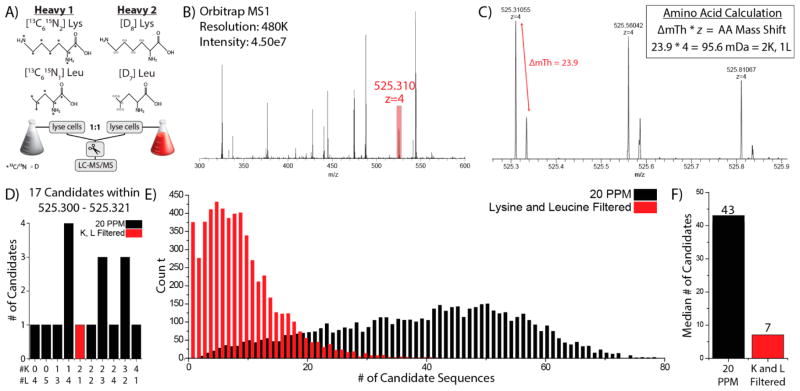
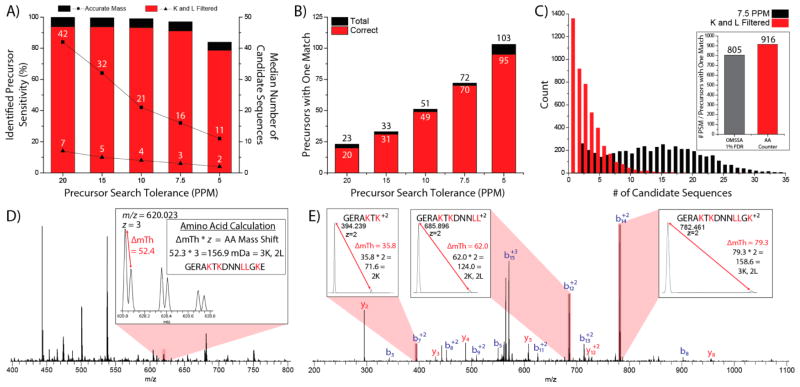
Metabolic labeling of cells using heavy amino acids is most commonly used for relative quantitation; however, partner mass shifts also detail the number of heavy amino acids contained within the precursor species. Here, we use a recently developed metabolic labeling technique, NeuCode (neutron encoding) stable isotope labeling with amino acids in cell culture (SILAC), which produces precursor partners spaced ~40 mDa apart to enable amino acid counting. We implement large scale counting of amino acids through a program, "Amino Acid Counter", which determines the most likely combination of amino acids within a precursor based on NeuCode SILAC partner spacing and filters candidate peptide sequences during a database search using this information. Counting the number of lysine residues for precursors selected for MS/MS decreases the median number of candidate sequences from 44 to 14 as compared to an accurate mass search alone (20 ppm). Furthermore, the ability to co-isolate and fragment NeuCode SILAC partners enables counting of lysines in product ions, and when the information is used, the median number of candidates is reduced to 7. We then demonstrate counting leucine in addition to lysine results in a 6-fold decrease in search space, 43 to 7, when compared to an accurate mass search. We use this scheme to analyze a nanoLC-MS/MS experiment and demonstrate that accurate mass plus lysine and leucine counting reduces the number of candidate sequences to one for ~20% of all precursors selected, demonstrating an ability to identify precursors without MS/MS analysis.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Coon JJ, Syka JEP, Shabanowitz J, et al. Biotechniques. 2005;38(4):519. - PubMed
-
- Eng JK, McCormack AL, Yates JR., III J Am Soc Mass Spectrom. 1994;5(11):976–989. - PubMed
-
- Perkins DN, Pappin DJC, Creasy DM, et al. Electrophoresis. 1999;20(18):3551–3567. - PubMed
-
- Craig R, Beavis RC. Bioinformatics. 2004;20(9):1466–1467. - PubMed
-
- Geer LY, Markey SP, Kowalak JA, et al. J Proteome Res. 2004;3(5):958–964. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
