

Hepatocytes produce TNF-α following hypoxia-reoxygenation and liver ischemia-reperfusion in a NADPH oxidase- and c-Src-dependent manner
- PMID: 23639811
- PMCID: PMC3725690
- DOI: 10.1152/ajpgi.00430.2012
Hepatocytes produce TNF-α following hypoxia-reoxygenation and liver ischemia-reperfusion in a NADPH oxidase- and c-Src-dependent manner
Abstract
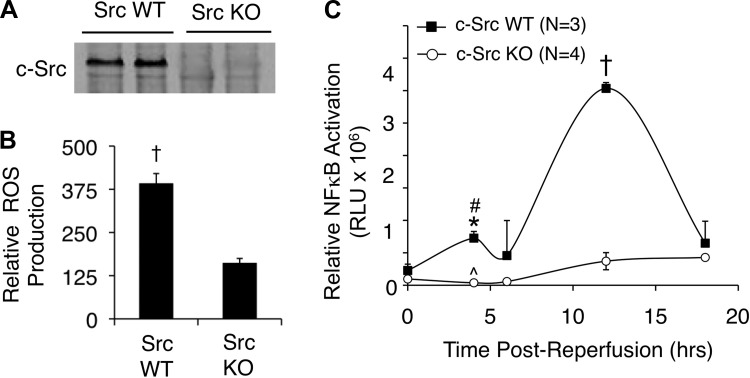
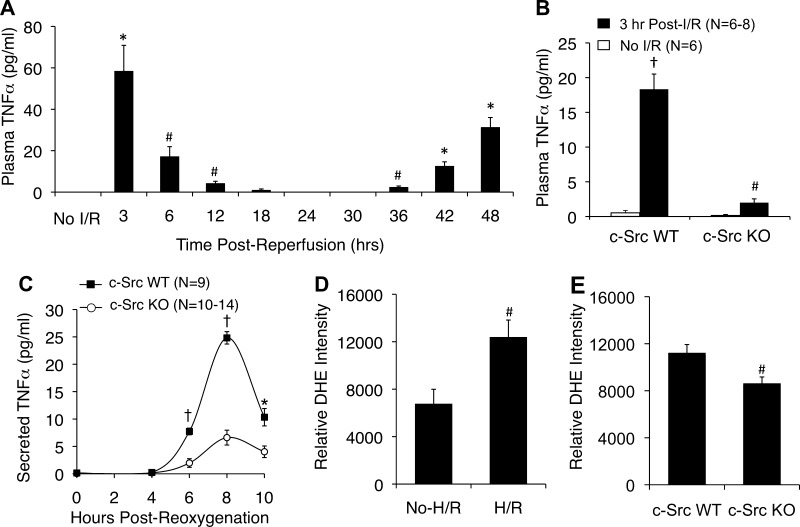
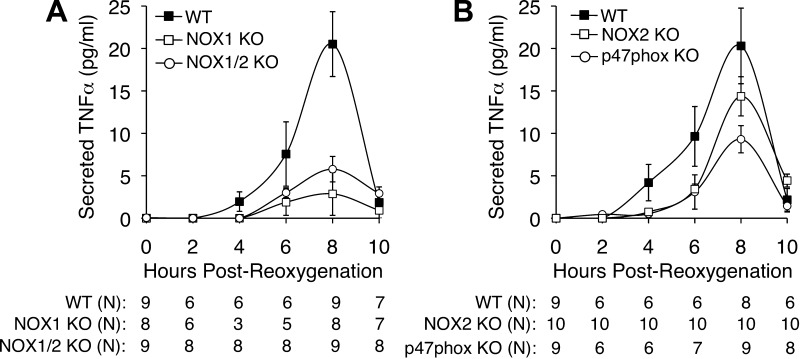
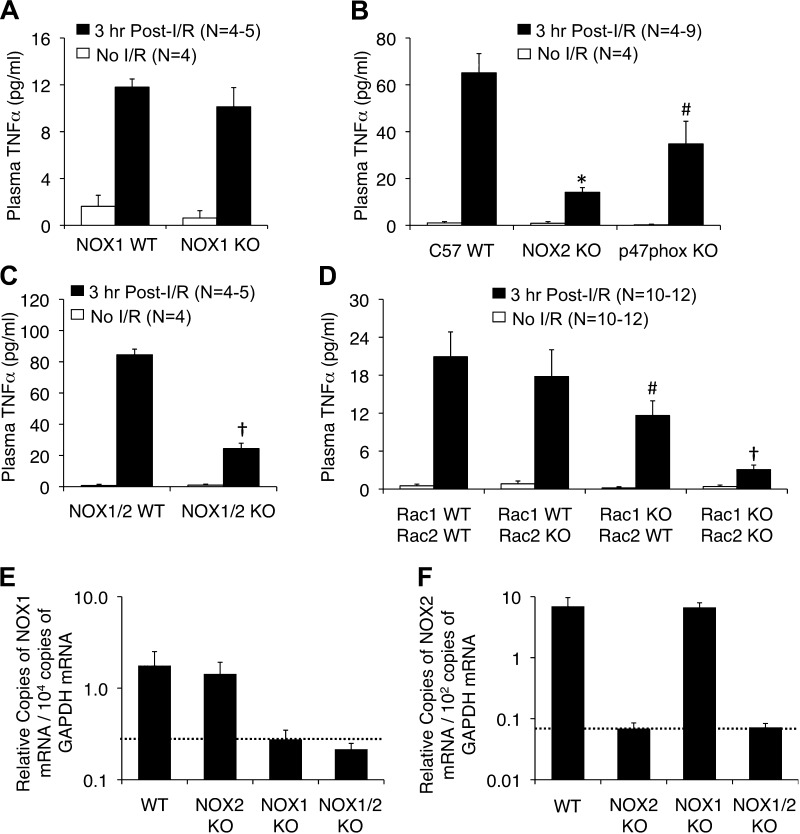
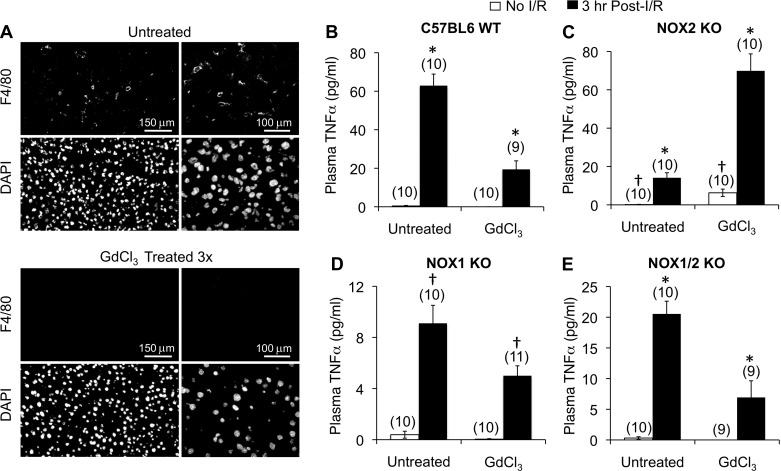
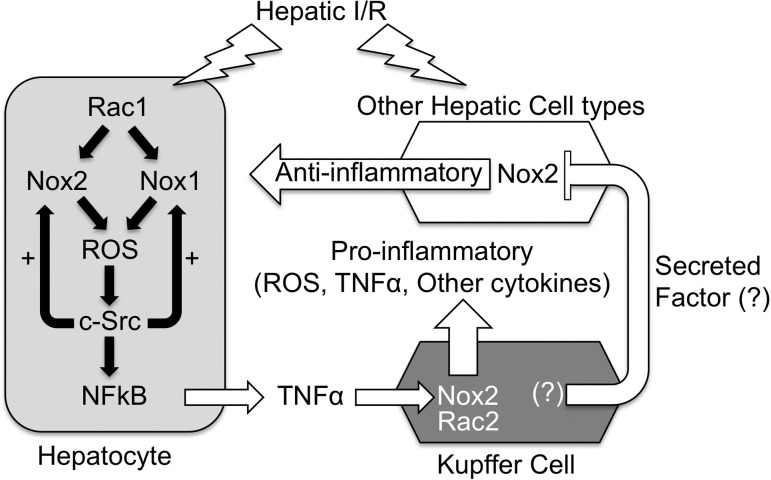
Cell line studies have previously demonstrated that hypoxia-reoxygenation (H/R) leads to the production of NADPH oxidase 1 and 2 (NOX1 and NOX2)-dependent reactive oxygen species (ROS) required for the activation of c-Src and NF-κB. We now extend these studies into mouse models to evaluate the contribution of hepatocytes to the NOX- and c-Src-dependent TNF-α production that follows H/R in primary hepatocytes and liver ischemia-reperfusion (I/R). In vitro, c-Src-deficient primary hepatocytes produced less ROS and TNF-α following H/R compared with controls. In vivo, c-Src-KO mice also had impaired TNF-α and NF-κB responses following partial lobar liver I/R. Studies in NOX1 and p47phox knockout primary hepatocytes demonstrated that both NOX1 and p47phox are partially required for H/R-mediated TNF-α production. To further investigate the involvement of NADPH oxidases in the production of TNF-α following liver I/R, we performed additional in vivo experiments in knockout mice deficient for NOX1, NOX2, p47phox, Rac1, and/or Rac2. Cumulatively, these results demonstrate that NOX2 and its activator subunits (p47phox and Rac) control the secretion of TNF-α by the liver following I/R. Interestingly, in the absence of Kupffer cells and NOX2, NOX1 played a dominant role in TNF-α production following hepatic I/R. However, NOX1 deletion alone had little effect on I/R-induced TNF-α. Thus Kupffer cell-derived factors and NOX2 act to suppress hepatic NOX1-dependent TNF-α production. We conclude that c-Src and NADPH oxidase components are necessary for redox-mediated production of TNF-α following liver I/R and that hepatocytes play an important role in this process.
Keywords: NADPH oxidase; c-Src; hepatocyte; redox; reperfusion injury.
Figures
Similar articles
-
Endosomal NADPH oxidase regulates c-Src activation following hypoxia/reoxygenation injury.Biochem J. 2008 May 1;411(3):531-41. doi: 10.1042/BJ20071534. Biochem J. 2008. PMID: 18397177 Free PMC article.
-
The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species generation mediated by NADPH oxidase-1.Mol Biol Cell. 2008 Jul;19(7):2984-94. doi: 10.1091/mbc.e08-02-0138. Epub 2008 May 7. Mol Biol Cell. 2008. PMID: 18463161 Free PMC article.
-
Andrographolide Antagonizes TNF-α-Induced IL-8 via Inhibition of NADPH Oxidase/ROS/NF-κB and Src/MAPKs/AP-1 Axis in Human Colorectal Cancer HCT116 Cells.J Agric Food Chem. 2018 May 23;66(20):5139-5148. doi: 10.1021/acs.jafc.8b00810. Epub 2018 May 14. J Agric Food Chem. 2018. PMID: 29672044
-
Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy.Cancer Lett. 2008 Jul 18;266(1):37-52. doi: 10.1016/j.canlet.2008.02.044. Epub 2008 Apr 10. Cancer Lett. 2008. PMID: 18406051 Free PMC article. Review.
-
Role of NADPH oxidases in liver fibrosis.Antioxid Redox Signal. 2014 Jun 10;20(17):2854-72. doi: 10.1089/ars.2013.5619. Epub 2014 Jan 24. Antioxid Redox Signal. 2014. PMID: 24040957 Free PMC article. Review.
Cited by
-
Pregnane X receptor modulates the inflammatory response in primary cultures of hepatocytes.Drug Metab Dispos. 2015 Mar;43(3):335-43. doi: 10.1124/dmd.114.062307. Epub 2014 Dec 19. Drug Metab Dispos. 2015. PMID: 25527709 Free PMC article.
-
Hyperglycemia Aggravates Hepatic Ischemia Reperfusion Injury by Inducing Chronic Oxidative Stress and Inflammation.Oxid Med Cell Longev. 2016;2016:3919627. doi: 10.1155/2016/3919627. Epub 2016 Aug 31. Oxid Med Cell Longev. 2016. PMID: 27656261 Free PMC article.
-
Effect of Metformin on Myocardial Injury Induced by Hepatic Ischemia-Reperfusion in Rats.Front Pharmacol. 2022 Apr 1;13:822743. doi: 10.3389/fphar.2022.822743. eCollection 2022. Front Pharmacol. 2022. PMID: 35431970 Free PMC article.
-
Cell-Specific Regulation of Inflammatory Cytokines and Acute-Phase Proteins by the Glucocorticoid Receptor.Mediators Inflamm. 2023 Nov 28;2023:4399998. doi: 10.1155/2023/4399998. eCollection 2023. Mediators Inflamm. 2023. PMID: 39619227 Free PMC article.
-
Aggravation of post-ischemic liver injury by overexpression of insulin-like growth factor binding protein 3.Sci Rep. 2015 Jun 15;5:11231. doi: 10.1038/srep11231. Sci Rep. 2015. PMID: 26073647 Free PMC article.
References
-
- Abe J, Takahashi M, Ishida M, Lee JD, Berk BC. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J Biol Chem 272: 20389– 20394, 1997 - PubMed
-
- Bhogal RH, Curbishley SM, Weston CJ, Adams DH, Afford SC. Reactive oxygen species mediate human hepatocyte injury during hypoxia/reoxygenation. Liver Transpl 16: 1303– 1313, 2010 - PubMed
-
- Boros P, Bromberg JS. New cellular and molecular immune pathways in ischemia-reperfusion injury. Am J Transplant 6: 652– 658, 2006 - PubMed
-
- Chen YX, Sato M, Kawachi K, Abe Y. Neutrophil-mediated liver injury during hepatic ischemia-reperfusion in rats. Hepatobiliary Pancreat Dis Int 5: 436– 442, 2006 - PubMed
-
- Diaz-Cruz A, Vilchis-Landeros MM, Guinzberg R, Villalobos-Molina R, Pina E. NOX2 activated by α1-adrenoceptors modulates hepatic metabolic routes stimulated by β-adrenoceptors. Free Radic Res 45: 1366– 1378, 2011 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous