

Fibroblast growth factor receptor like-1 (FGFRL1) interacts with SHP-1 phosphatase at insulin secretory granules and induces beta-cell ERK1/2 protein activation
- PMID: 23640895
- PMCID: PMC3682584
- DOI: 10.1074/jbc.M112.440677
Fibroblast growth factor receptor like-1 (FGFRL1) interacts with SHP-1 phosphatase at insulin secretory granules and induces beta-cell ERK1/2 protein activation
Abstract
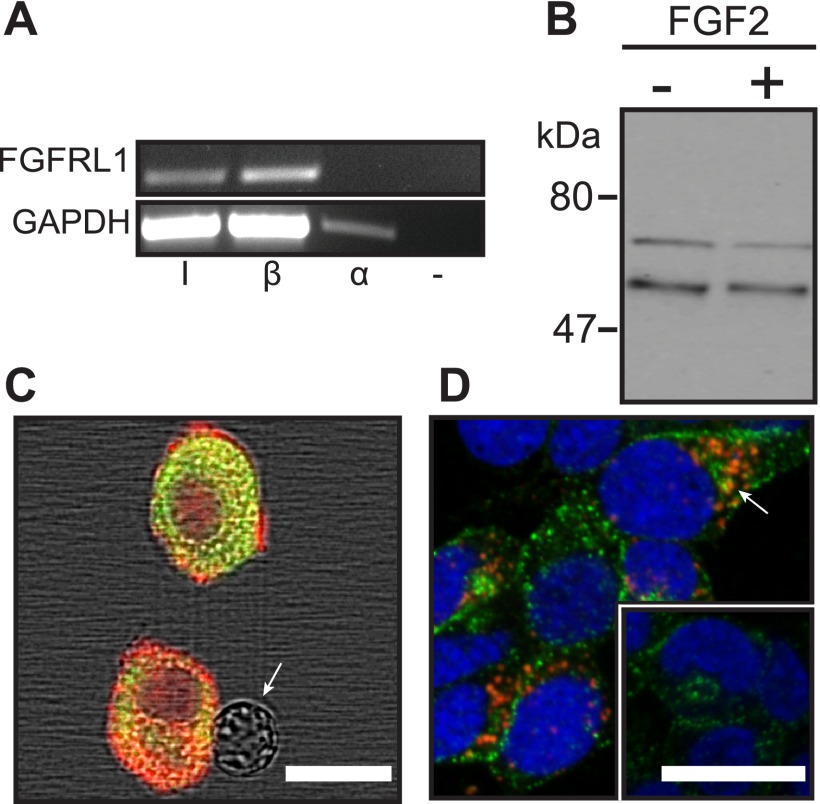
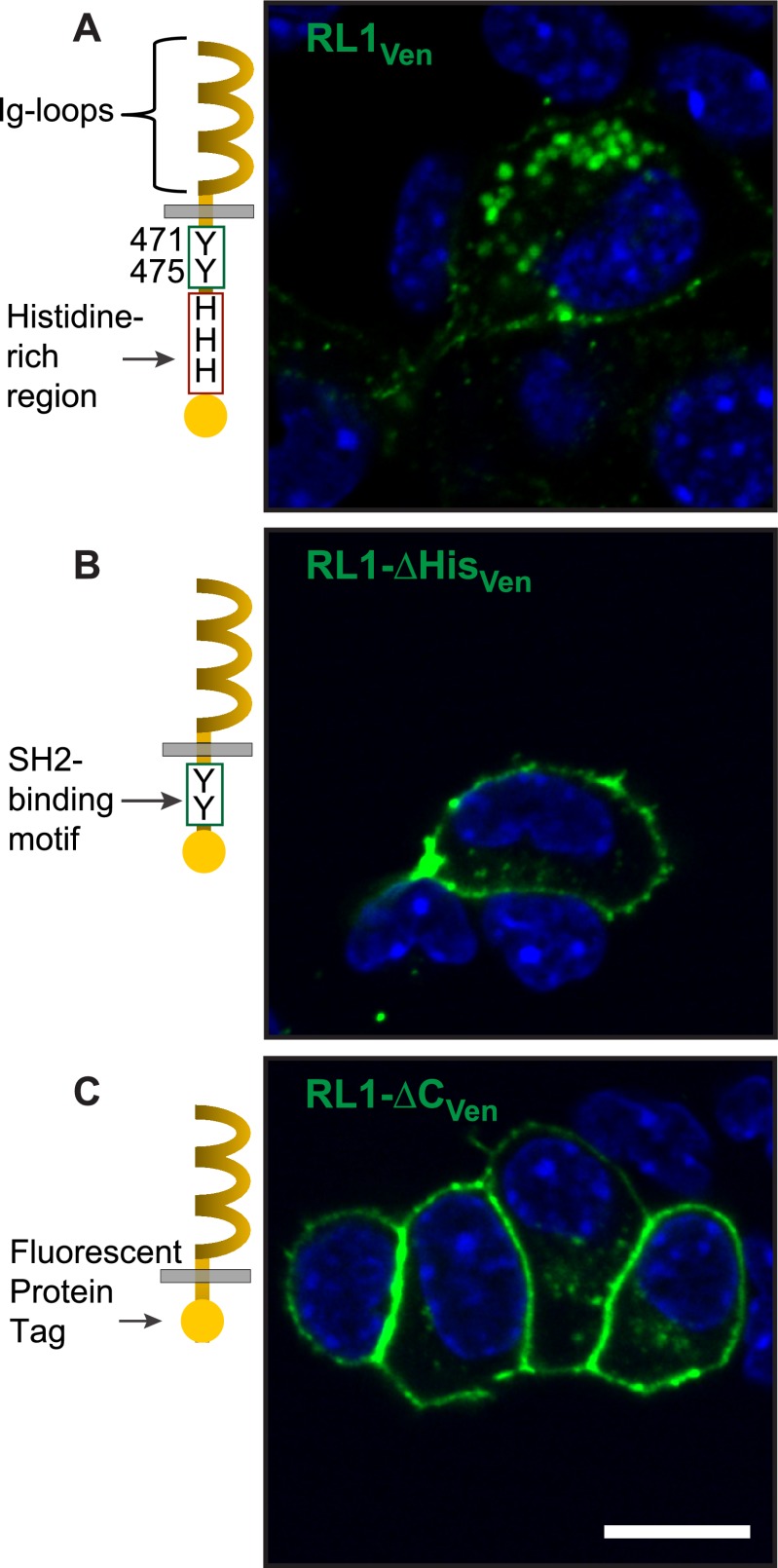
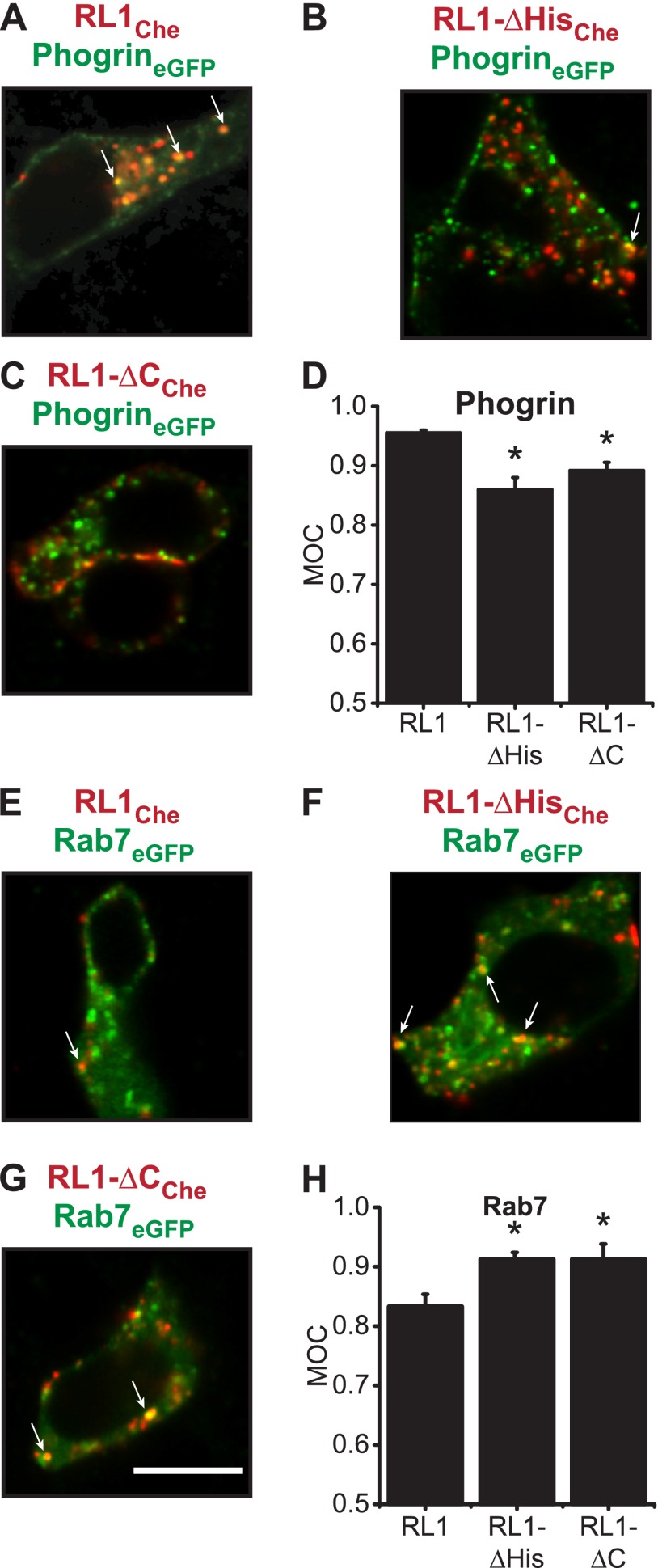
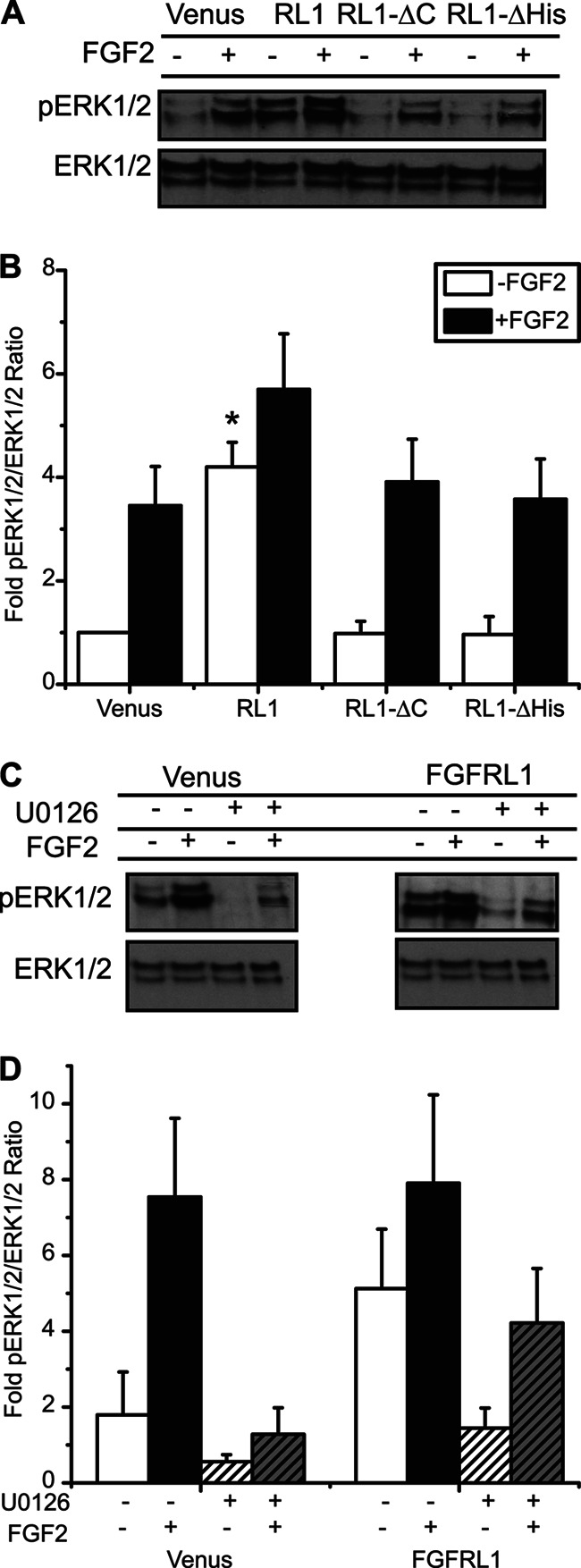
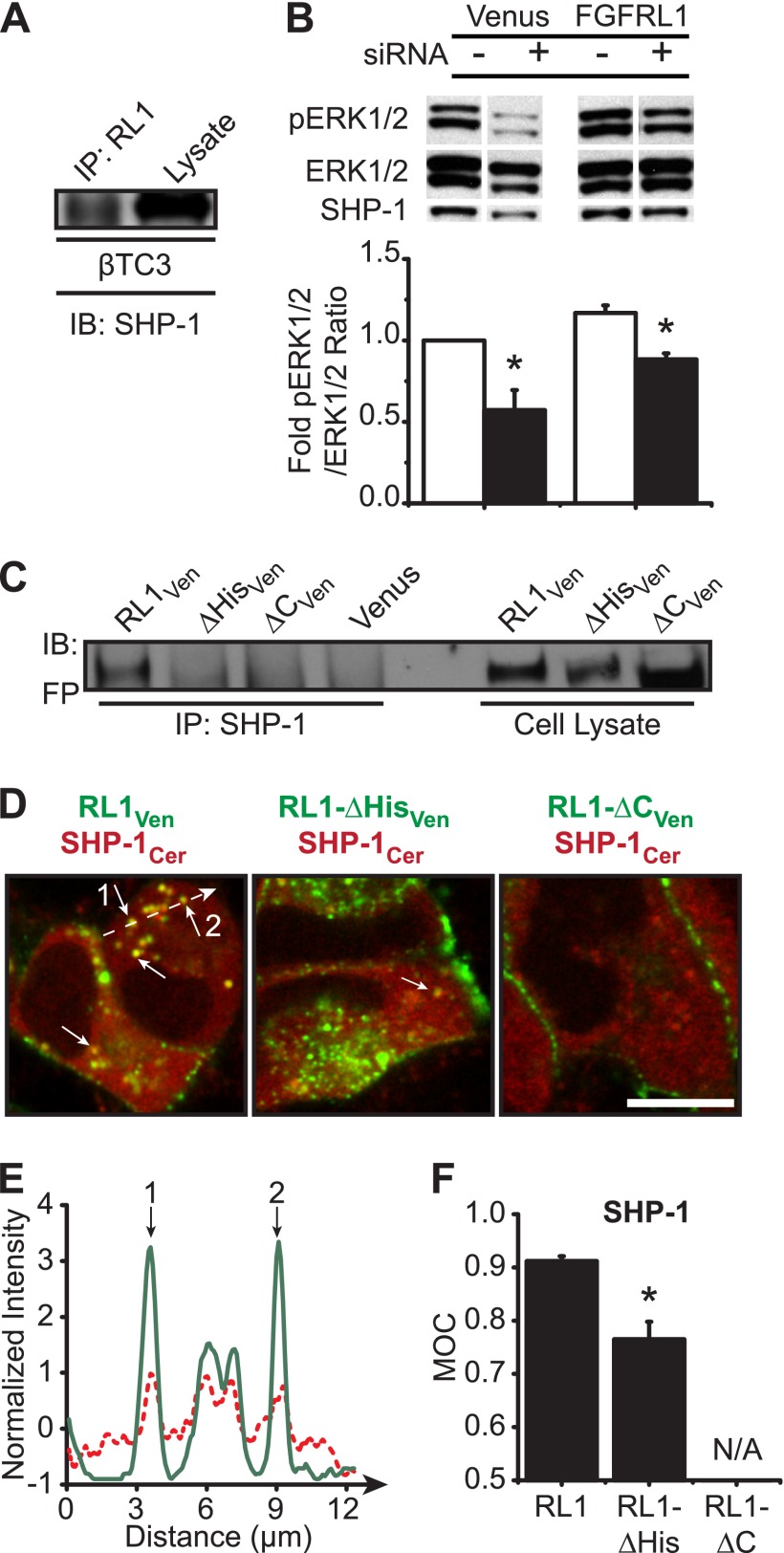
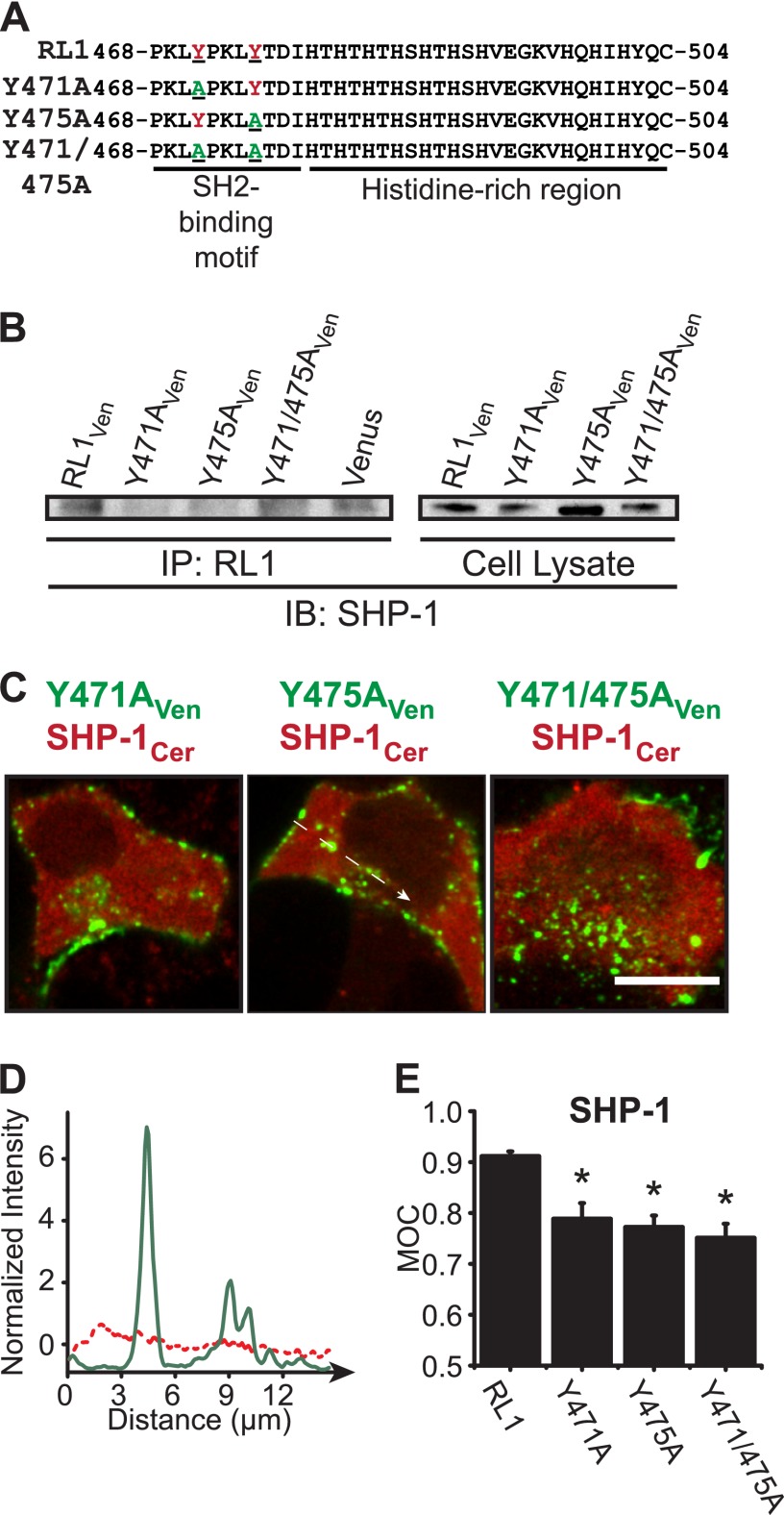
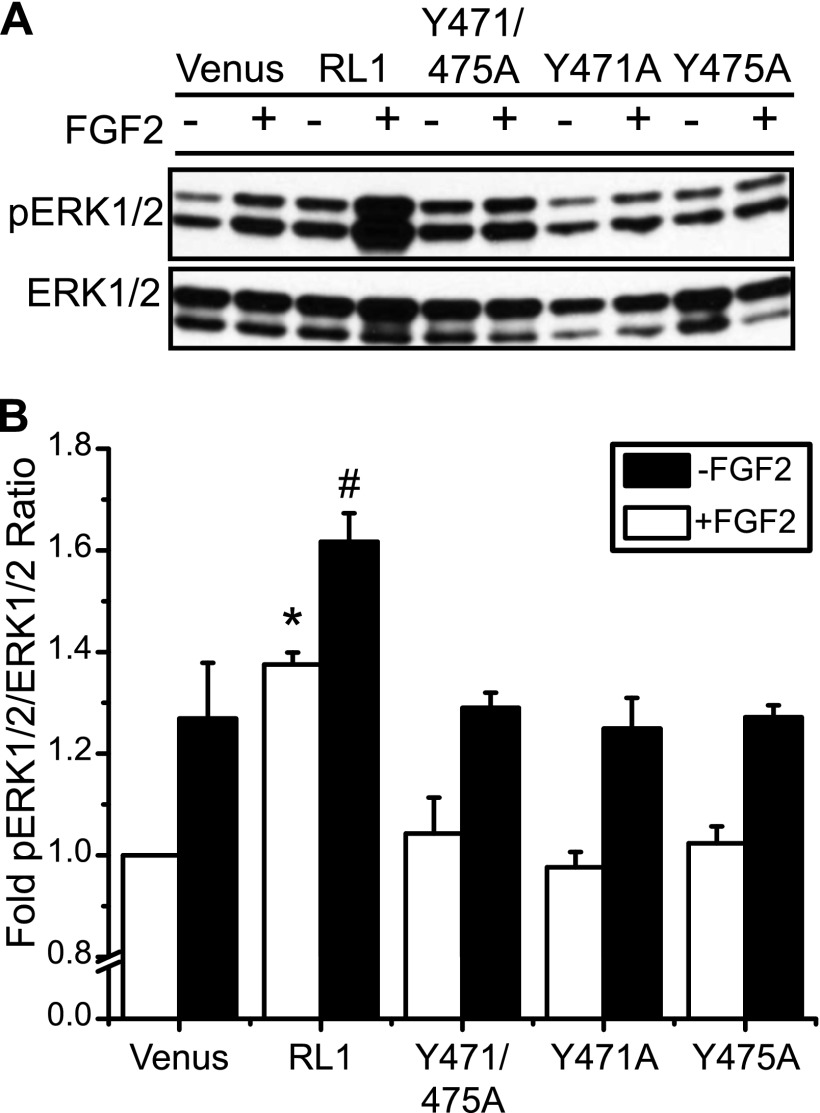
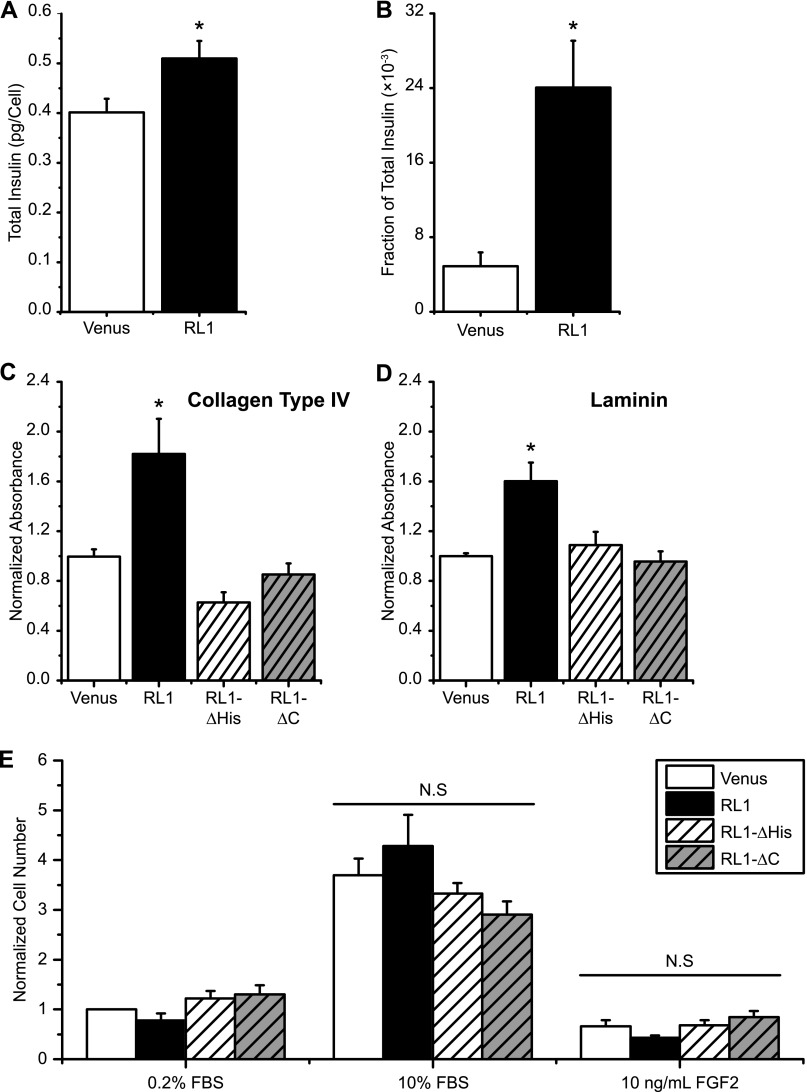
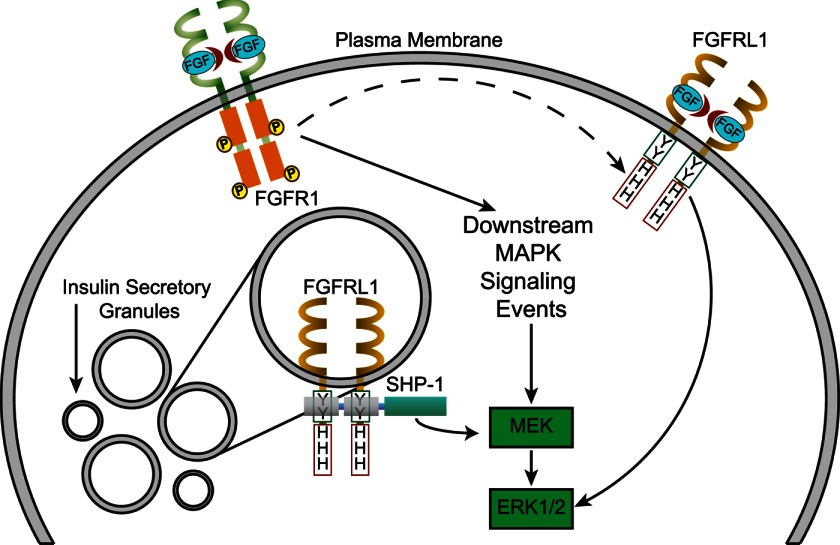
FGFRL1 is a newly identified member of the fibroblast growth factor receptor (FGFR) family expressed in adult pancreas. Unlike canonical FGFRs that initiate signaling via tyrosine kinase domains, the short intracellular sequence of FGFRL1 consists of a putative Src homology domain-2 (SH2)-binding motif adjacent to a histidine-rich C terminus. As a consequence of nonexistent kinase domains, FGFRL1 has been postulated to act as a decoy receptor to inhibit canonical FGFR ligand-induced signaling. In pancreatic islet beta-cells, canonical FGFR1 signaling affects metabolism and insulin processing. This study determined beta-cell expression of FGFRL1 as well as consequent effects on FGFR1 signaling and biological responses. We confirmed FGFRL1 expression at the plasma membrane and within distinct intracellular granules of both primary beta-cells and βTC3 cells. Fluorescent protein-tagged FGFRL1 (RL1) induced a significant ligand-independent increase in MAPK signaling. Removal of the histidine-rich domain (RL1-ΔHis) or entire intracellular sequence (RL1-ΔC) resulted in greater retention at the plasma membrane and significantly reduced ligand-independent ERK1/2 responses. The SHP-1 phosphatase was identified as an RL1-binding substrate. Point mutation of the SH2-binding motif reduced the ability of FGFRL1 to bind SHP-1 and activate ERK1/2 but did not affect receptor localization to insulin secretory granules. Finally, overexpression of RL1 increased cellular insulin content and matrix adhesion. Overall, these data suggest that FGFRL1 does not function as a decoy receptor in beta-cells, but rather it enhances ERK1/2 signaling through association of SHP-1 with the receptor's intracellular SH2-binding motif.
Keywords: Beta-cell; FGFR5; FGFRL1; Fibroblast Growth Factor Receptor (FGFR); MAP Kinases (MAPKs); PTPN6; Receptor Tyrosine Kinase; SHP-1; Signal Transduction.
Figures
References
-
- Hart A. W., Baeza N., Apelqvist A., Edlund H. (2000) Attenuation of FGF signalling in mouse beta-cells leads to diabetes. Nature 408, 864–868 - PubMed
-
- Arany E., Hill D. J. (2000) Ontogeny of fibroblast growth factors in the early development of the rat endocrine pancreas. Pediatr. Res. 48, 389–403 - PubMed
-
- Basilico C., Moscatelli D. (1992) The FGF family of growth factors and oncogenes Adv. Cancer Res. 59, 115–165 - PubMed
-
- Powers C. J., McLeskey S. W., Wellstein A. (2000) Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 7, 165–197 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
