

Dnmt2-dependent methylomes lack defined DNA methylation patterns
- PMID: 23641003
- PMCID: PMC3666705
- DOI: 10.1073/pnas.1306723110
Dnmt2-dependent methylomes lack defined DNA methylation patterns
Abstract
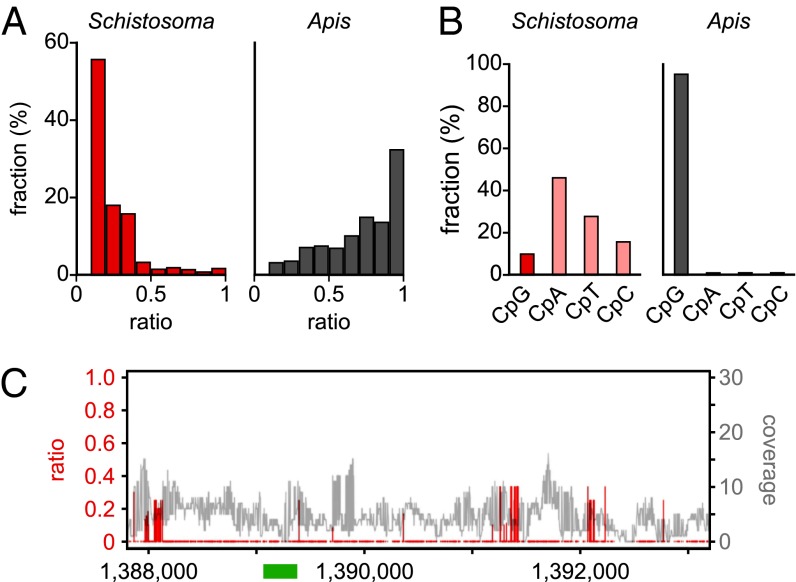
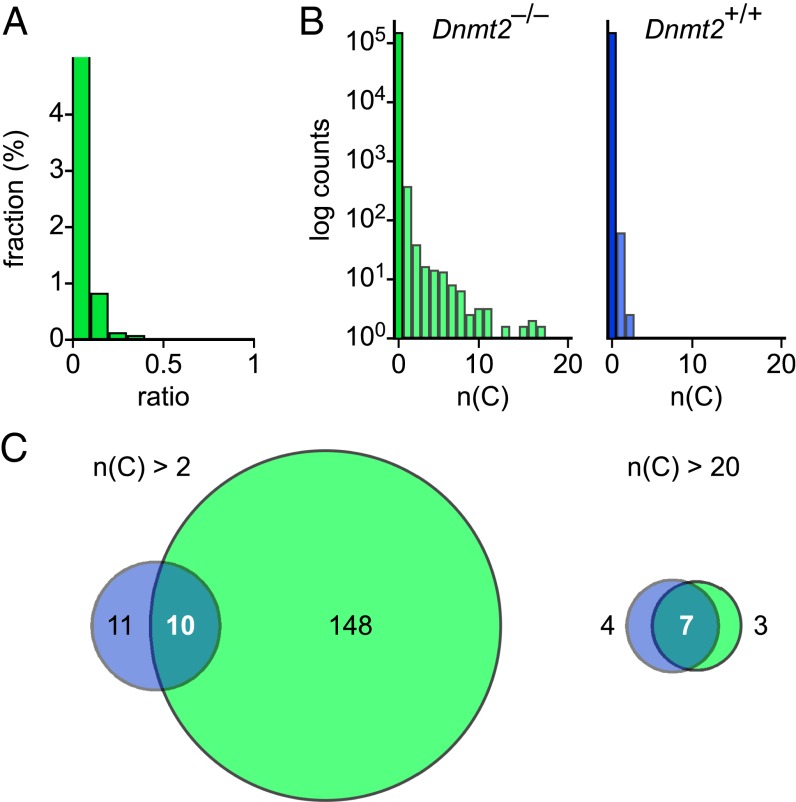
Several organisms have retained methyltransferase 2 (Dnmt2) as their only candidate DNA methyltransferase gene. However, information about Dnmt2-dependent methylation patterns has been limited to a few isolated loci and the results have been discussed controversially. In addition, recent studies have shown that Dnmt2 functions as a tRNA methyltransferase, which raised the possibility that Dnmt2-only genomes might be unmethylated. We have now used whole-genome bisulfite sequencing to analyze the methylomes of Dnmt2-only organisms at single-base resolution. Our results show that the genomes of Schistosoma mansoni and Drosophila melanogaster lack detectable DNA methylation patterns. Residual unconverted cytosine residues shared many attributes with bisulfite deamination artifacts and were observed at comparable levels in Dnmt2-deficient flies. Furthermore, genetically modified Dnmt2-only mouse embryonic stem cells lost the DNA methylation patterns found in wild-type cells. Our results thus uncover fundamental differences among animal methylomes and suggest that DNA methylation is dispensable for a considerable number of eukaryotic organisms.
Keywords: RNA methylation; epigenetics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Mohn F, Schübeler D. Genetics and epigenetics: Stability and plasticity during cellular differentiation. Trends Genet. 2009;25(3):129–136. - PubMed
-
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–440. - PubMed
-
- Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. - PubMed
-
- Yoder JA, Bestor TH. A candidate mammalian DNA methyltransferase related to pmt1p of fission yeast. Hum Mol Genet. 1998;7(2):279–284. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
