

Quantum mechanics/molecular mechanics modeling of regioselectivity of drug metabolism in cytochrome P450 2C9
- PMID: 23641937
- PMCID: PMC3670427
- DOI: 10.1021/ja402016p
Quantum mechanics/molecular mechanics modeling of regioselectivity of drug metabolism in cytochrome P450 2C9
Abstract
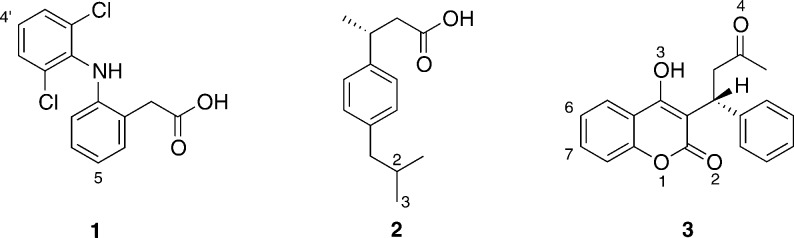
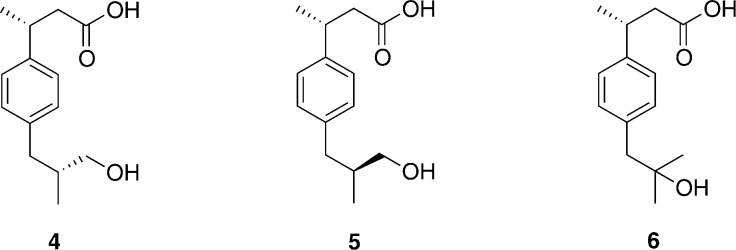
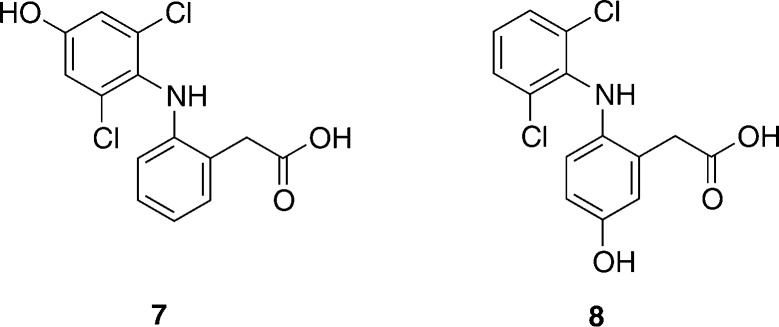
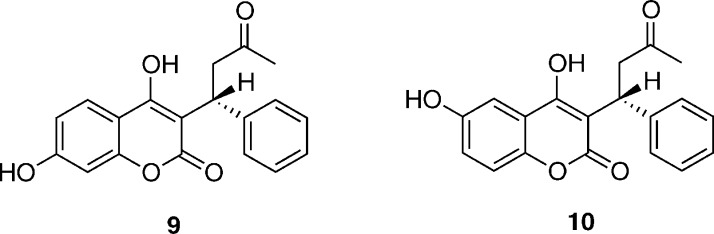
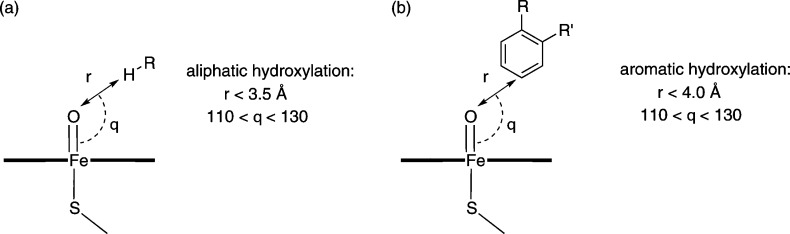
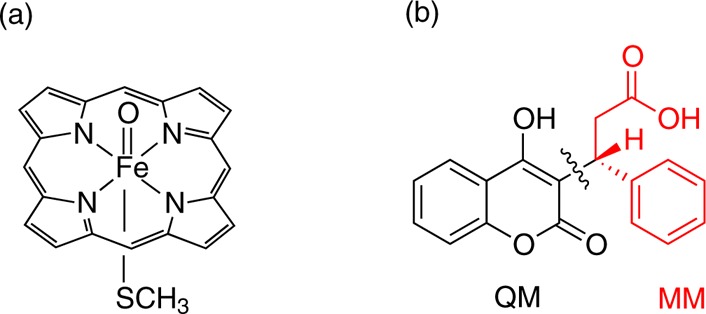
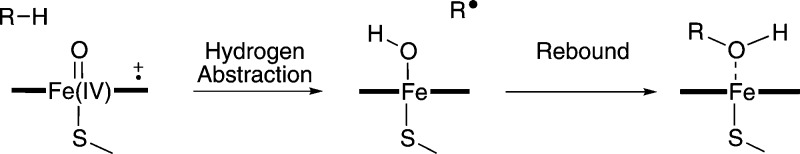
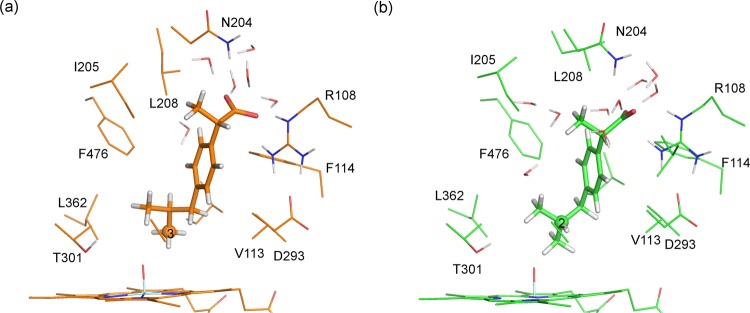
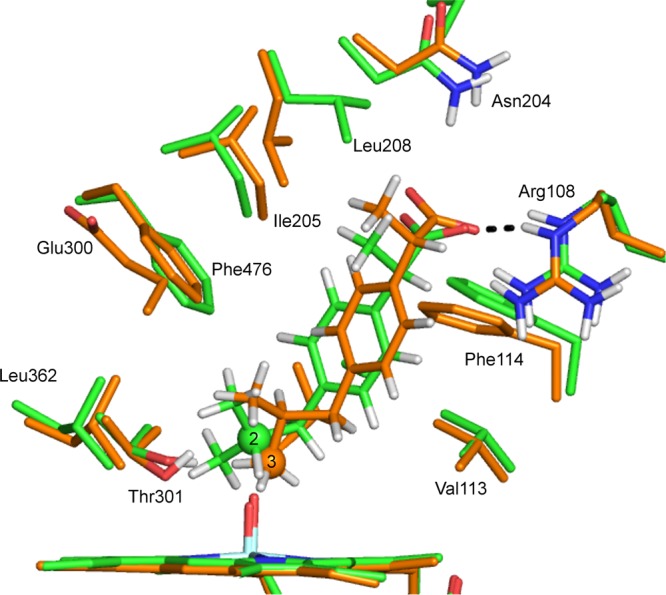
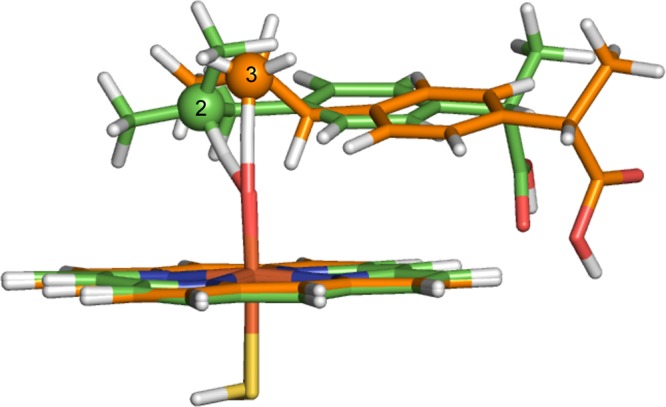
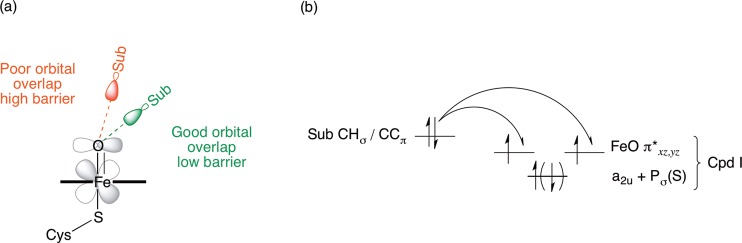
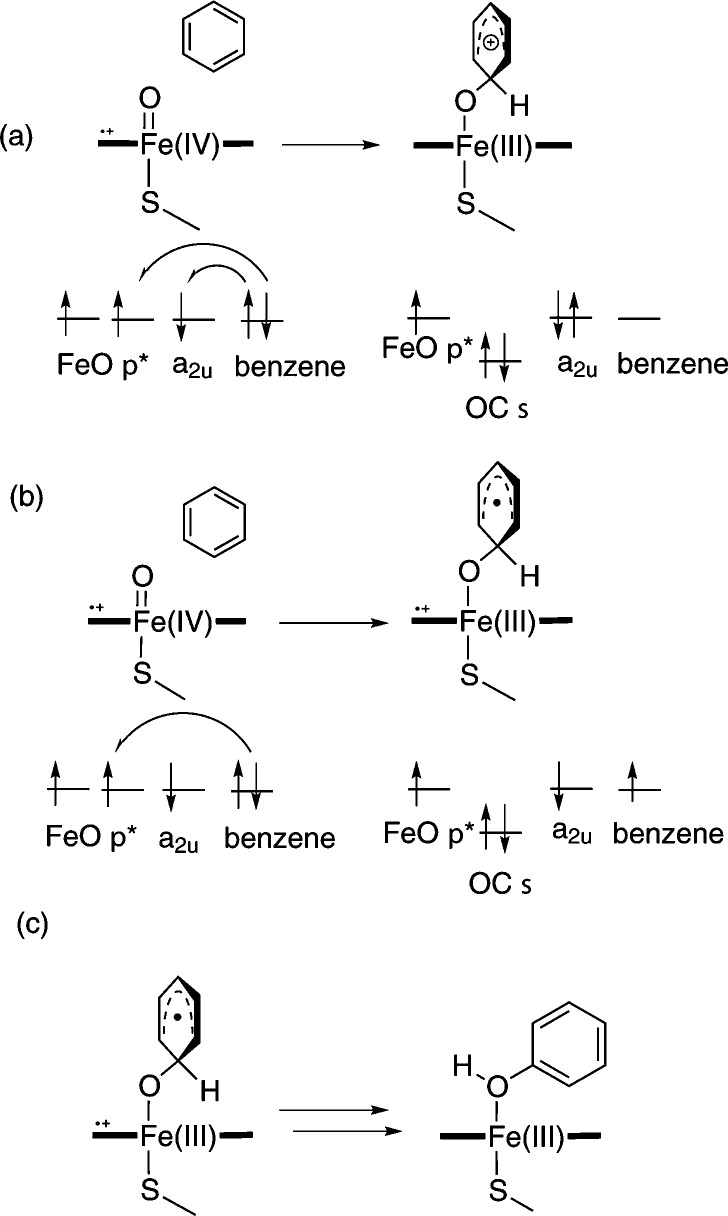
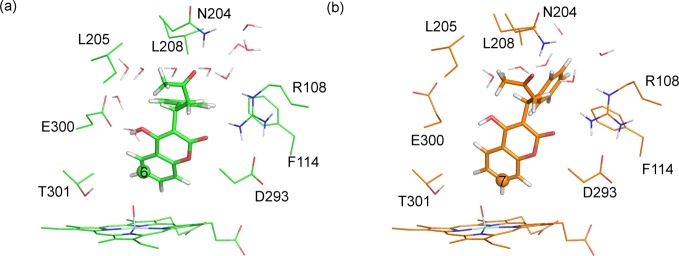
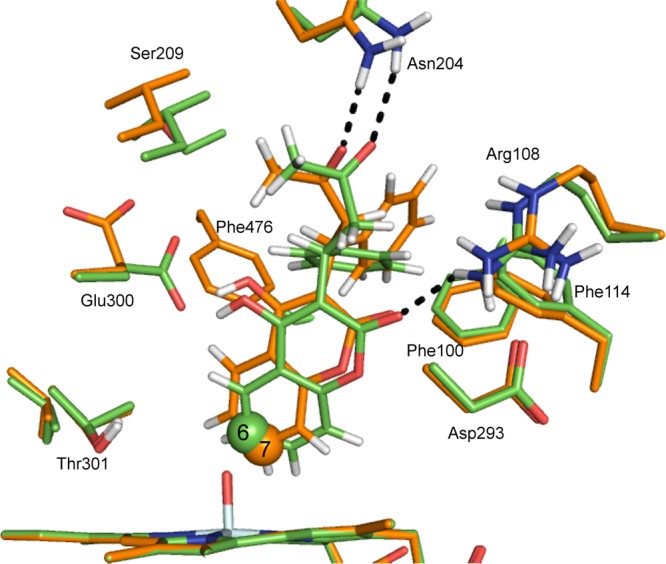
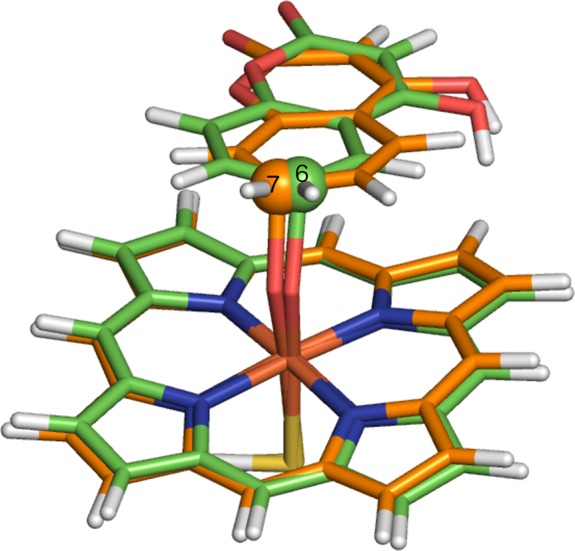
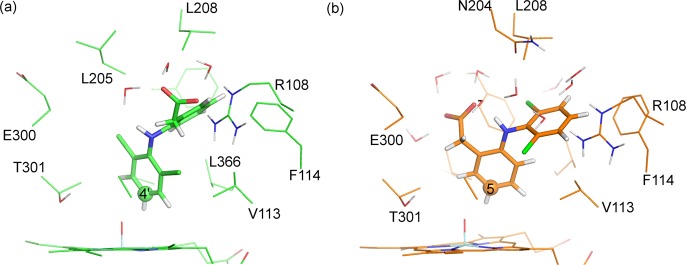
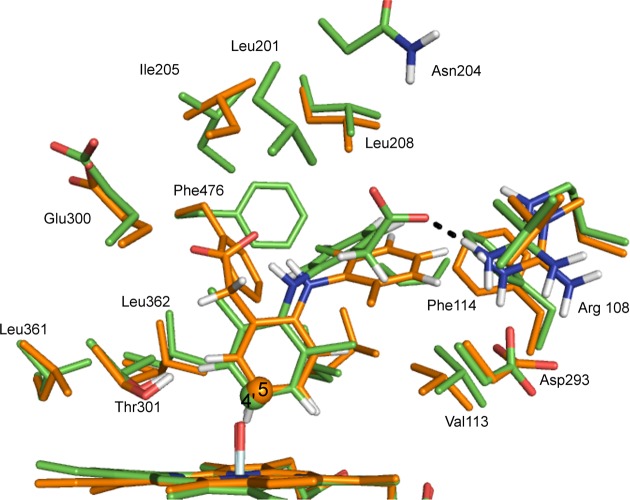
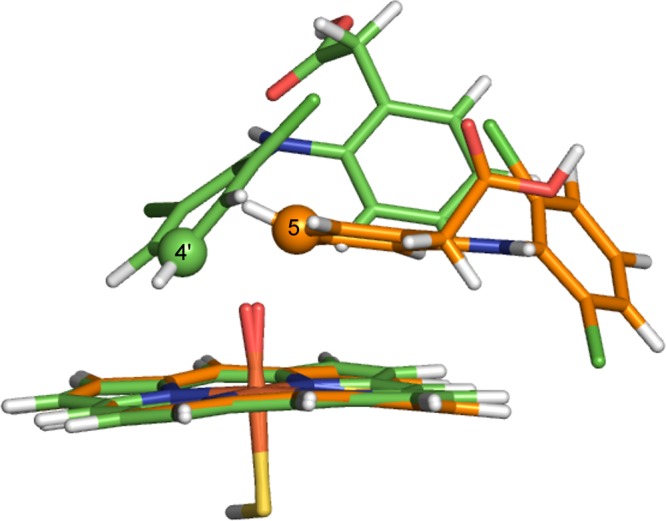
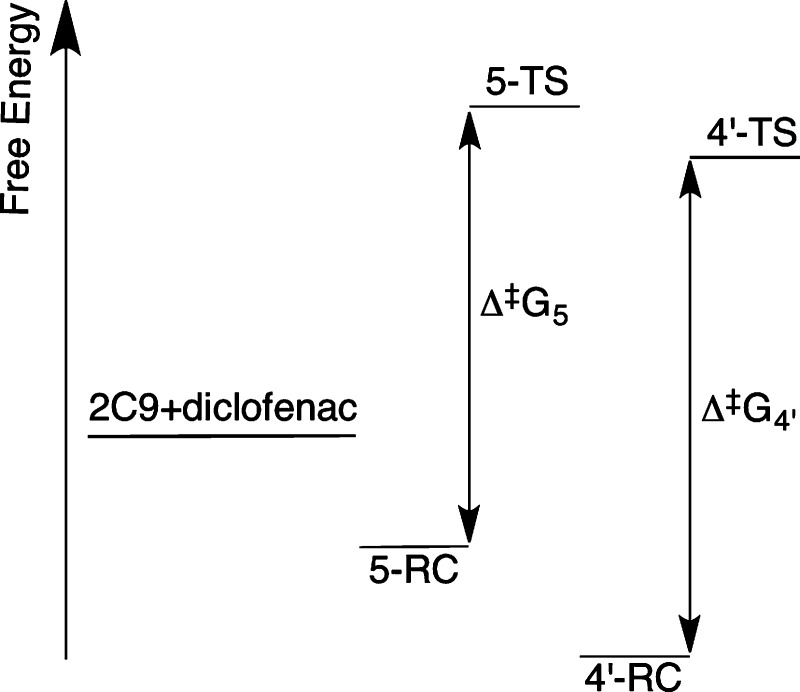
Cytochrome P450 enzymes (P450s) are important in drug metabolism and have been linked to adverse drug reactions. P450s display broad substrate reactivity, and prediction of metabolites is complex. QM/MM studies of P450 reactivity have provided insight into important details of the reaction mechanisms and have the potential to make predictions of metabolite formation. Here we present a comprehensive study of the oxidation of three widely used pharmaceutical compounds (S-ibuprofen, diclofenac, and S-warfarin) by one of the major drug-metabolizing P450 isoforms, CYP2C9. The reaction barriers to substrate oxidation by the iron-oxo species (Compound I) have been calculated at the B3LYP-D/CHARMM27 level for different possible metabolism sites for each drug, on multiple pathways. In the cases of ibuprofen and warfarin, the process with the lowest activation energy is consistent with the experimentally preferred metabolite. For diclofenac, the pathway leading to the experimentally observed metabolite is not the one with the lowest activation energy. This apparent inconsistency with experiment might be explained by the two very different binding modes involved in oxidation at the two competing positions. The carboxylate of diclofenac interacts strongly with the CYP2C9 Arg108 side chain in the transition state for formation of the observed metabolite-but not in that for the competing pathway. We compare reaction barriers calculated both in the presence and in the absence of the protein and observe a marked improvement in selectivity prediction ability upon inclusion of the protein for all of the substrates studied. The barriers calculated with the protein are generally higher than those calculated in the gas phase. This suggests that active-site residues surrounding the substrate play an important role in controlling selectivity in CYP2C9. The results show that inclusion of sampling (particularly) and dispersion effects is important in making accurate predictions of drug metabolism selectivity of P450s using QM/MM methods.
Figures
References
-
- Ortiz de Montellano P. R., Ed. Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, 2005.
-
- Lonsdale R.; Harvey J. N.; Mulholland A. J. In Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; de Visser S. P., Kumar D., Eds.; RSC Publishing: Cambridge, UK, 2011; Chapter 11, pp 366–399.
-
- de Graaf C.; Vermeulen N. P. E.; Feenstra K. A. J. Med. Chem. 2005, 48, 2725–2755. - PubMed
-
- Bathelt C. M.; Ridder L.; Mulholland A. J.; Harvey J. N. Org. Biomol. Chem. 2004, 2, 2998–3005. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
