

Readout of epigenetic modifications
- PMID: 23642229
- PMCID: PMC4696766
- DOI: 10.1146/annurev-biochem-072711-165700
Readout of epigenetic modifications
Abstract
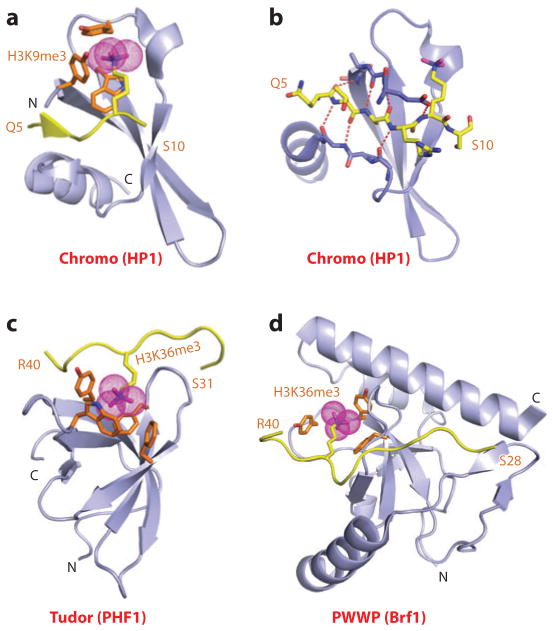
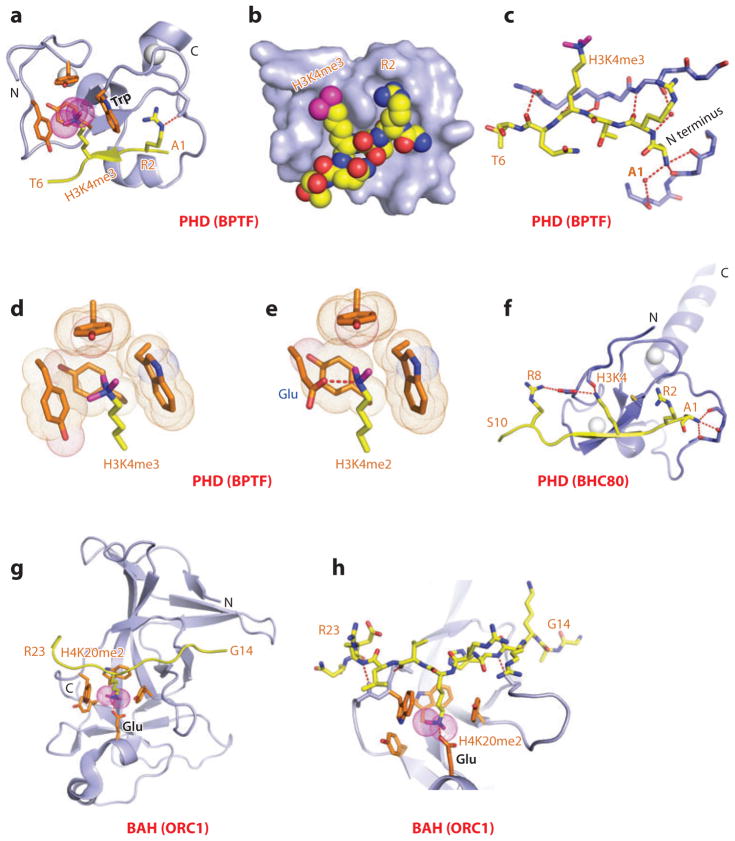
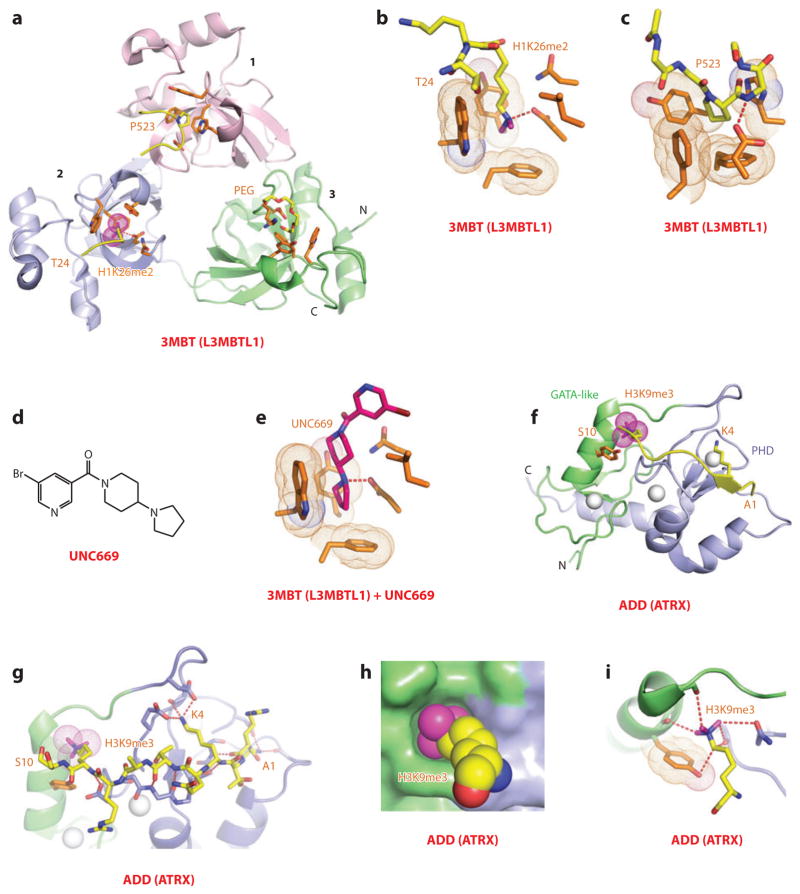
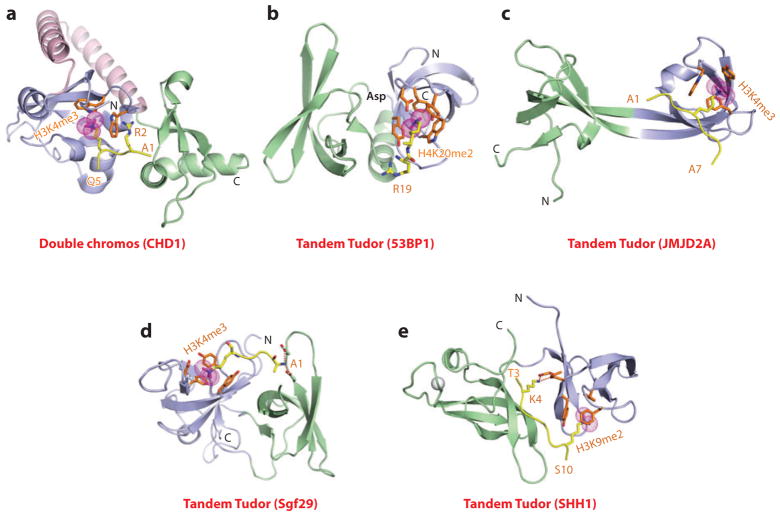
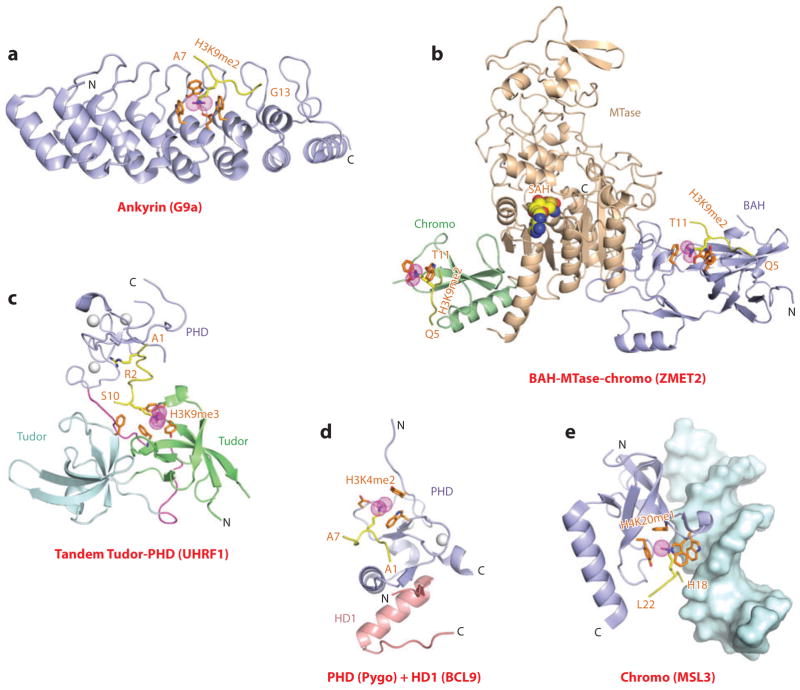
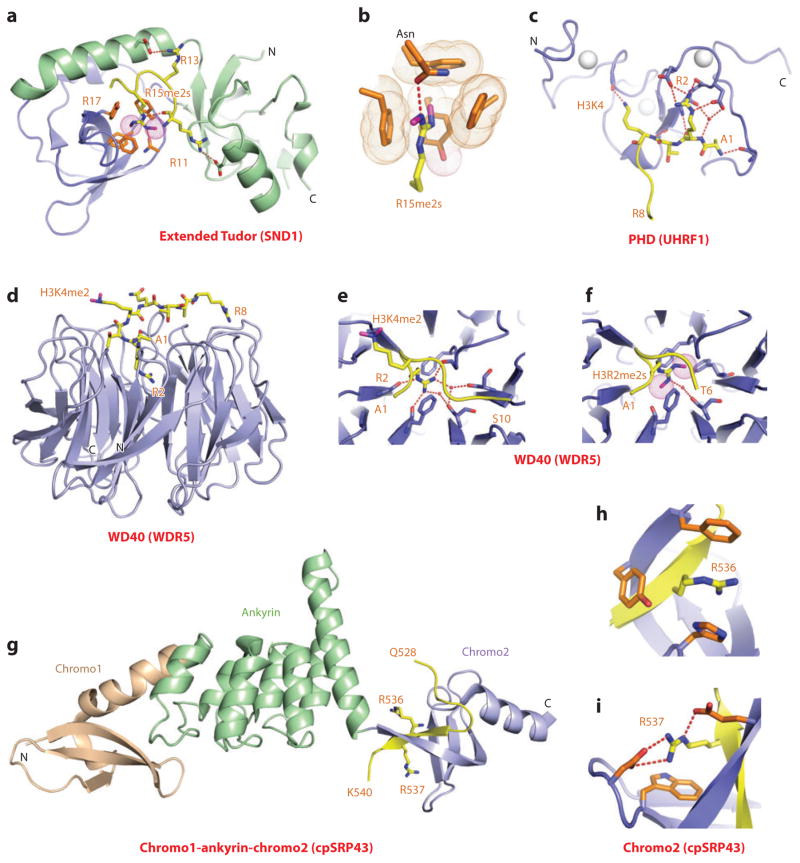
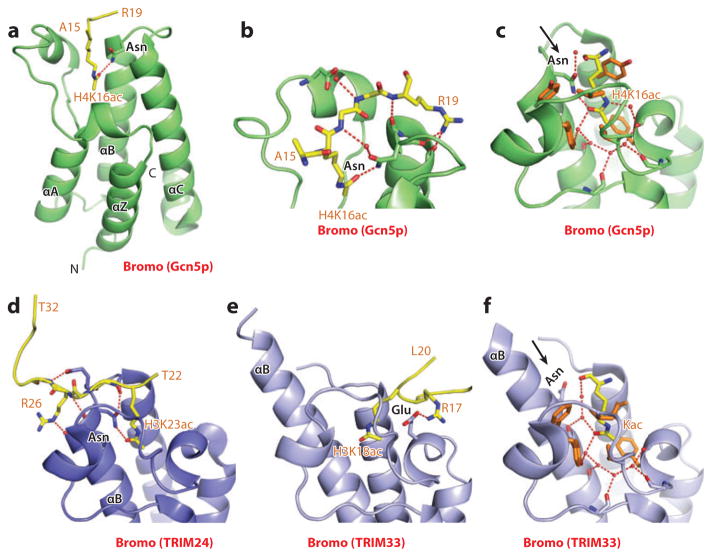
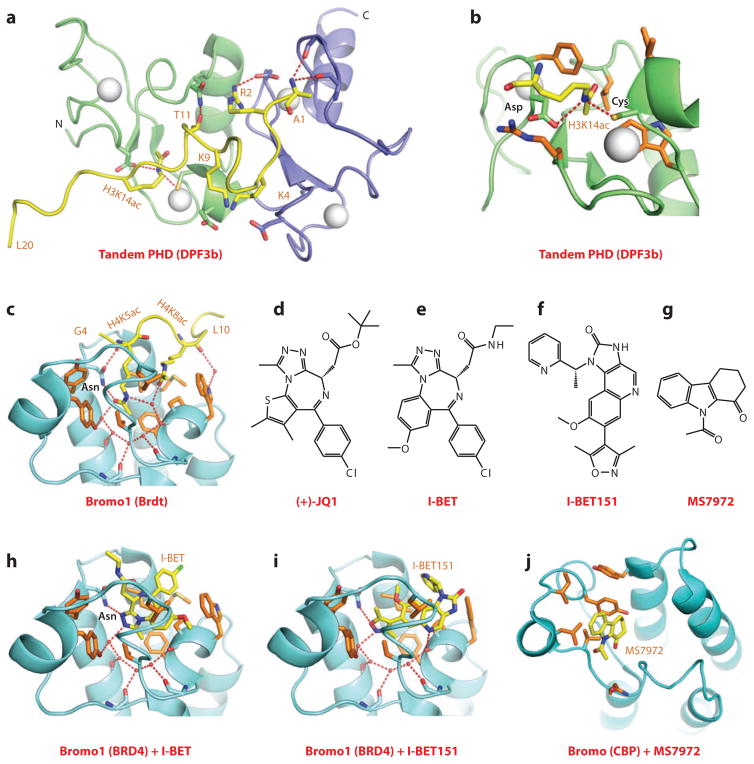
This review focuses on a structure-based analysis of histone posttranslational modification (PTM) readout, where the PTMs serve as docking sites for reader modules as part of larger complexes displaying chromatin modifier and remodeling activities, with the capacity to alter chromatin architecture and templated processes. Individual topics addressed include the diversity of reader-binding pocket architectures and common principles underlying readout of methyl-lysine and methyl-arginine marks, their unmodified counterparts, as well as acetyl-lysine and phosphoserine marks. The review also discusses the impact of multivalent readout of combinations of PTMs localized at specific genomic sites by linked binding modules on processes ranging from gene transcription to repair. Additional topics include cross talk between histone PTMs, histone mimics, epigenetic-based diseases, and drug-based therapeutic intervention. The review ends by highlighting new initiatives and advances, as well as future challenges, toward the promise of enhancing our structural and mechanistic understanding of the readout of histone PTMs at the nucleosomal level.
Figures
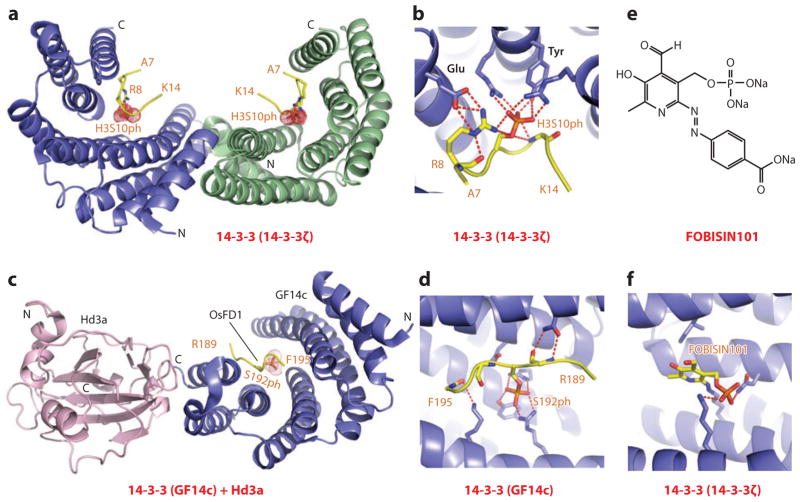
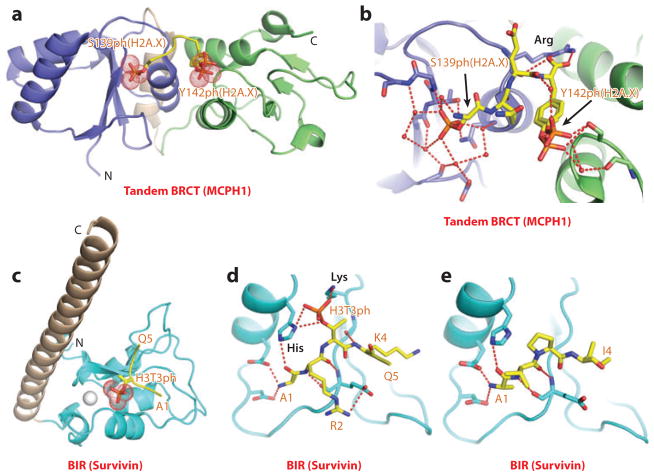
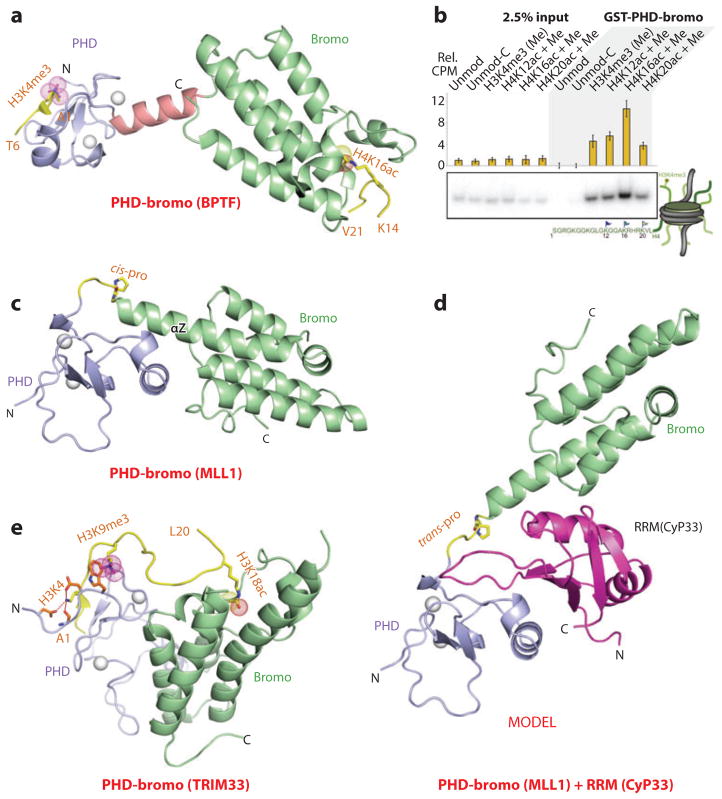
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
