

Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism
- PMID: 23643382
- PMCID: PMC3644636
- DOI: 10.1016/j.ajhg.2013.04.008
Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism
Abstract
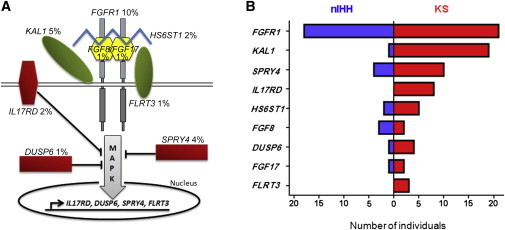
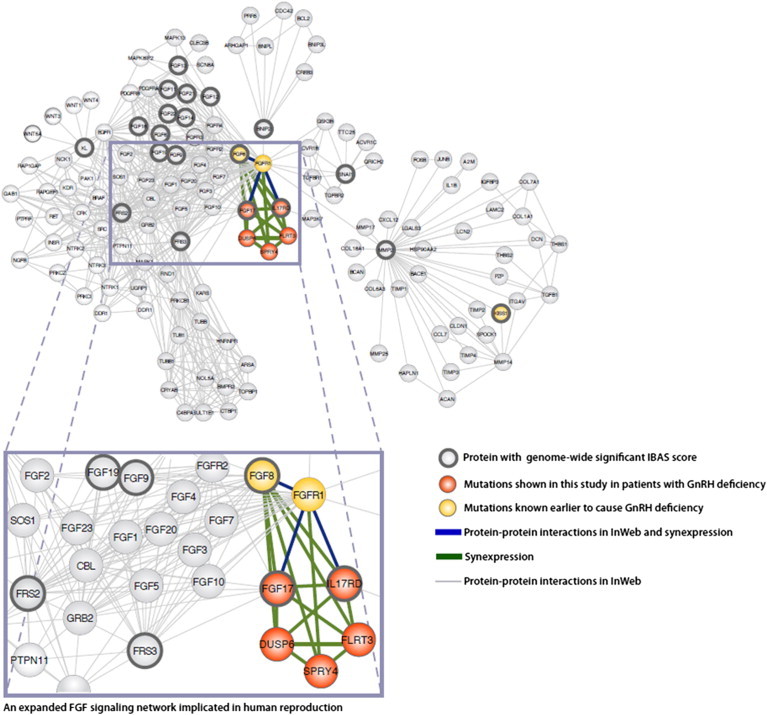
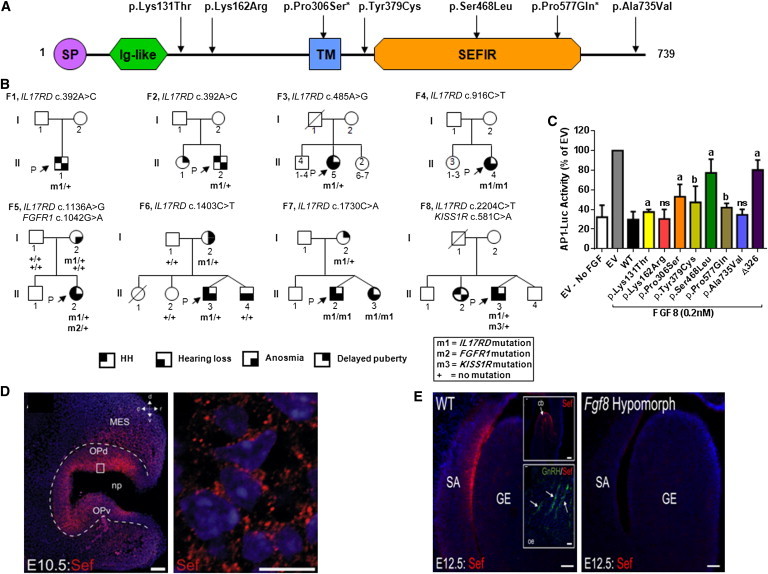
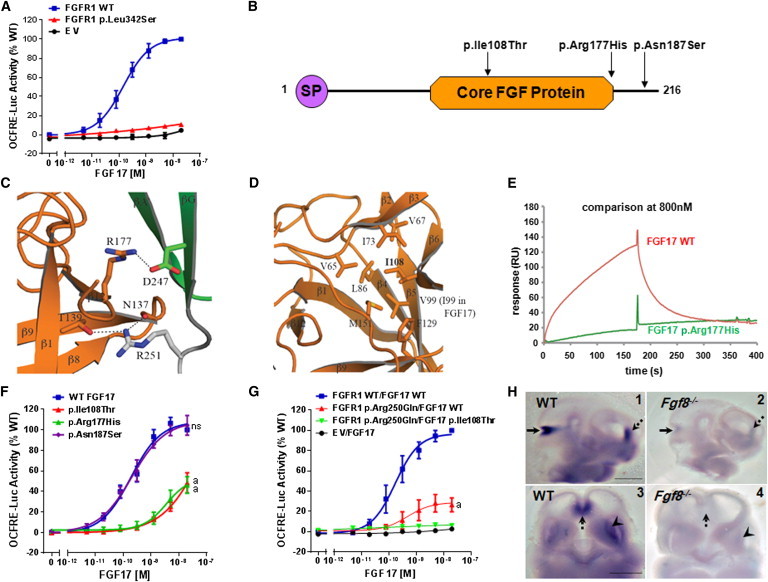
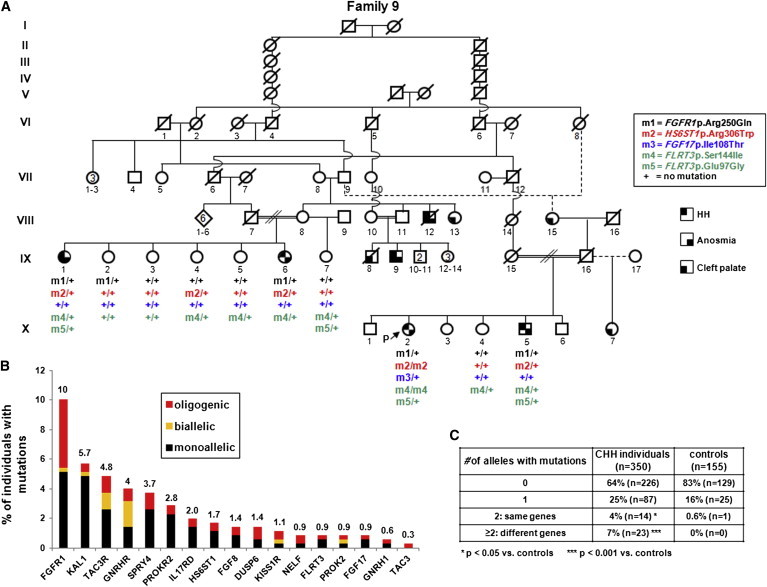
Congenital hypogonadotropic hypogonadism (CHH) and its anosmia-associated form (Kallmann syndrome [KS]) are genetically heterogeneous. Among the >15 genes implicated in these conditions, mutations in FGF8 and FGFR1 account for ~12% of cases; notably, KAL1 and HS6ST1 are also involved in FGFR1 signaling and can be mutated in CHH. We therefore hypothesized that mutations in genes encoding a broader range of modulators of the FGFR1 pathway might contribute to the genetics of CHH as causal or modifier mutations. Thus, we aimed to (1) investigate whether CHH individuals harbor mutations in members of the so-called "FGF8 synexpression" group and (2) validate the ability of a bioinformatics algorithm on the basis of protein-protein interactome data (interactome-based affiliation scoring [IBAS]) to identify high-quality candidate genes. On the basis of sequence homology, expression, and structural and functional data, seven genes were selected and sequenced in 386 unrelated CHH individuals and 155 controls. Except for FGF18 and SPRY2, all other genes were found to be mutated in CHH individuals: FGF17 (n = 3 individuals), IL17RD (n = 8), DUSP6 (n = 5), SPRY4 (n = 14), and FLRT3 (n = 3). Independently, IBAS predicted FGF17 and IL17RD as the two top candidates in the entire proteome on the basis of a statistical test of their protein-protein interaction patterns to proteins known to be altered in CHH. Most of the FGF17 and IL17RD mutations altered protein function in vitro. IL17RD mutations were found only in KS individuals and were strongly linked to hearing loss (6/8 individuals). Mutations in genes encoding components of the FGF pathway are associated with complex modes of CHH inheritance and act primarily as contributors to an oligogenic genetic architecture underlying CHH.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Quinton R., Duke V.M., Robertson A., Kirk J.M., Matfin G., de Zoysa P.A., Azcona C., MacColl G.S., Jacobs H.S., Conway G.S. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin. Endocrinol. (Oxf.) 2001;55:163–174. - PubMed
-
- Dodé C., Levilliers J., Dupont J.M., De Paepe A., Le Dû N., Soussi-Yanicostas N., Coimbra R.S., Delmaghani S., Compain-Nouaille S., Baverel F. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003;33:463–465. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- T32 HD007396/HD/NICHD NIH HHS/United States
- R01HD15788/HD/NICHD NIH HHS/United States
- 2R01DE013686-11/DE/NIDCR NIH HHS/United States
- U54HD028138/HD/NICHD NIH HHS/United States
- R01 NS034661/NS/NINDS NIH HHS/United States
- R01 HD056264/HD/NICHD NIH HHS/United States
- R01 HD043341/HD/NICHD NIH HHS/United States
- R01 DE013686/DE/NIDCR NIH HHS/United States
- R01 NS34661/NS/NINDS NIH HHS/United States
- R01 HD015788/HD/NICHD NIH HHS/United States
- R01 HD042634/HD/NICHD NIH HHS/United States
- P30 DK063720/DK/NIDDK NIH HHS/United States
- K24 HD067388/HD/NICHD NIH HHS/United States
- R01HD056264/HD/NICHD NIH HHS/United States
- U54 HD028138/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
