

Hydroxycarbamide: clinical aspects
- PMID: 23643402
- PMCID: PMC4629816
- DOI: 10.1016/j.crvi.2012.09.006
Hydroxycarbamide: clinical aspects
Abstract
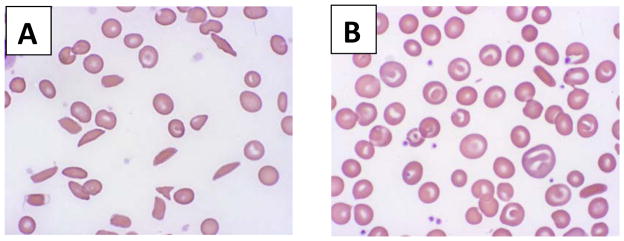
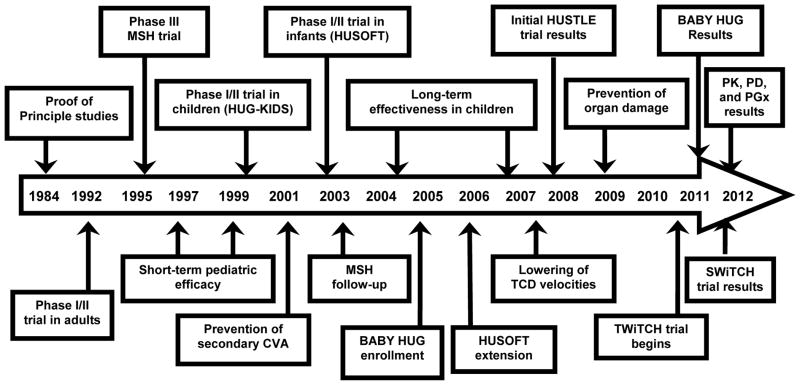
Due to its oral route of administration and mild toxicity profile, as well as its potent laboratory and clinical effects, hydroxyurea (or hydroxycarbamide) has been the primary focus of fetal hemoglobin (HbF) induction strategies for the treatment of children with sickle cell anemia (SCA). When administered orally once a day, hydroxyurea treatment is very well tolerated with little short-term toxicity. Hydroxyurea has documented laboratory efficacy with increases in Hb and HbF; treatment also significantly reduces the number of painful episodes, acute chest syndrome, transfusions, and hospitalizations. Most young patients reach a maximum tolerated dose of hydroxyurea at 25-30 mg/kg/d, where they will achieve key laboratory thresholds (Hb ≥ 9 g/dL and HbF ≥ 20%) without excessive myelosuppression. Potential long-term toxicities continue to be of great concern and should be monitored in all patients with SCA who receive hydroxyurea therapy. To date, however, no increases in stroke, myelodysplasia, or carcinogenicity have been detected in SCA patient cohorts, with drug exposure now reaching 15 years for some treated children. Taken together, available evidence suggests that hydroxyurea represents an inexpensive and effective treatment option that should be offered to most, if not all, patients with SCA. As countries in Africa develop newborn screening programs to identify SCA, the widespread use of hydroxyurea may prove to be a useful treatment to help ameliorate the disease in resource-limited settings. Hydroxyurea is the only currently available disease-modifying therapy for SCA, and is emerging as a safe and effective treatment for all patients with SCA, in both developed and developing countries.
Copyright © 2012 Académie des sciences. Published by Elsevier SAS. All rights reserved.
Figures
References
-
- Marcus SJ, Ware RE. Physiologic decline in fetal hemoglobin parameters in infants with sickle cell disease: implications for pharmacological intervention. J Pediatr Hematol/Oncol. 1999;21(5):407–411. - PubMed
-
- Brown AK, Sleeper LA, Miller ST, Pegelow CH, Gill FM, Waclawiw MA. Reference values and hematologic changes from birth to 5 years in patients with sickle cell disease. Cooperative Study of Sickle Cell Disease. Arch Pediatr Adolesc Med. 1994;148(8):796–804. - PubMed
-
- Powars D, Hiti A. Sickle cell anemia. Beta-S gene cluster haplotypes as genetic markers for severe disease expression. Am J Dis Child. 1993;147(11):1197–1202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials