

Population genomics of post-vaccine changes in pneumococcal epidemiology
- PMID: 23644493
- PMCID: PMC3725542
- DOI: 10.1038/ng.2625
Population genomics of post-vaccine changes in pneumococcal epidemiology
Abstract
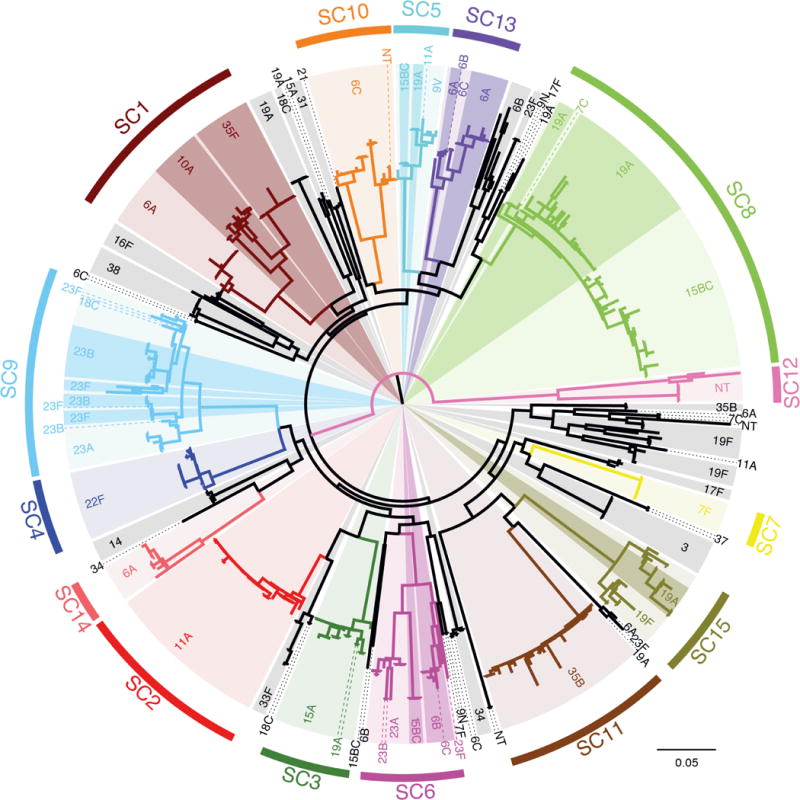
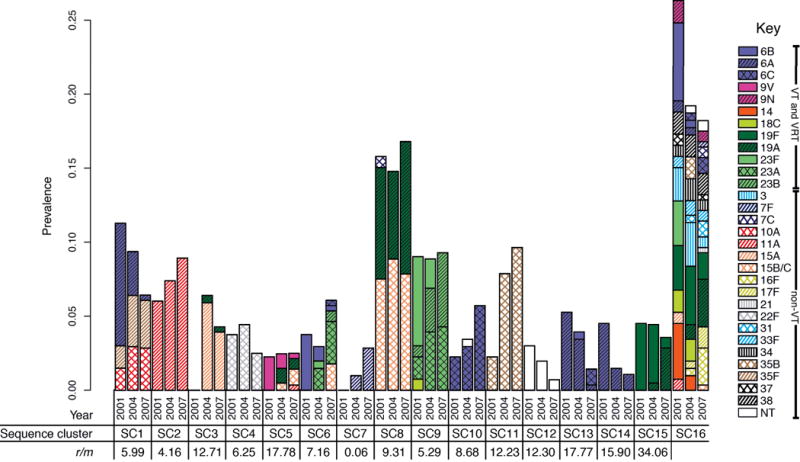
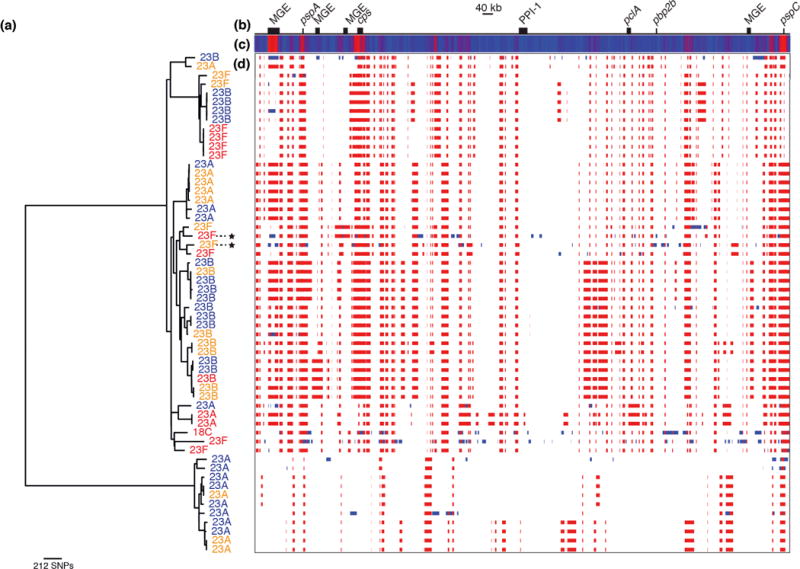
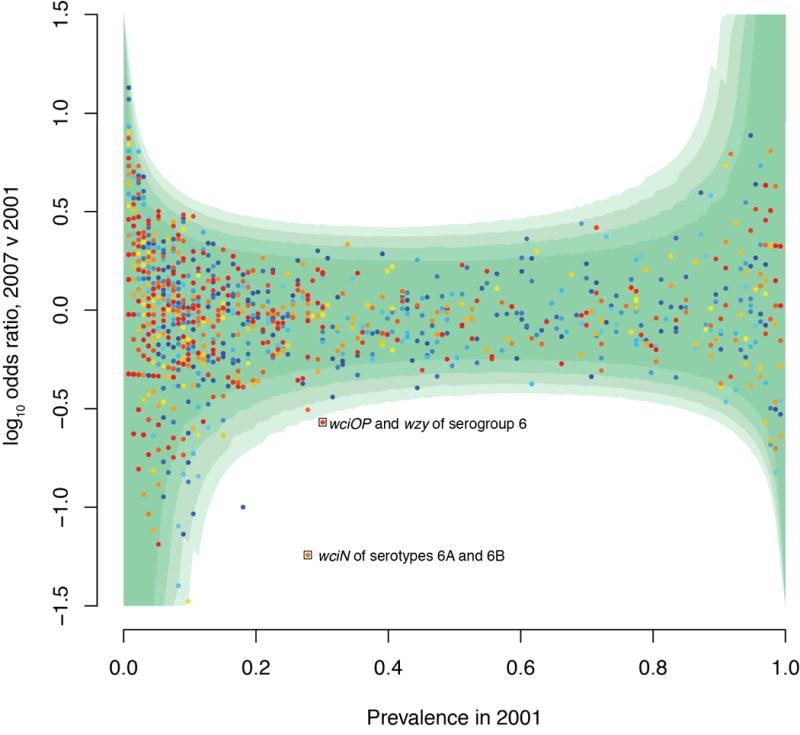
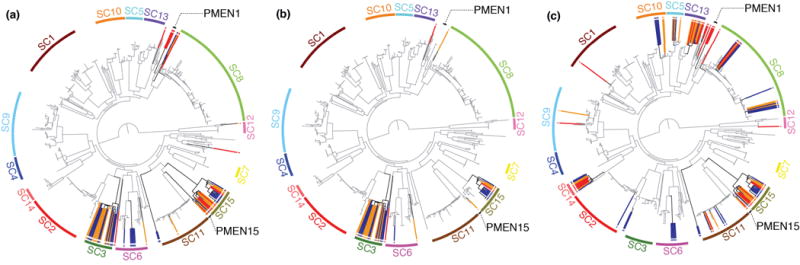
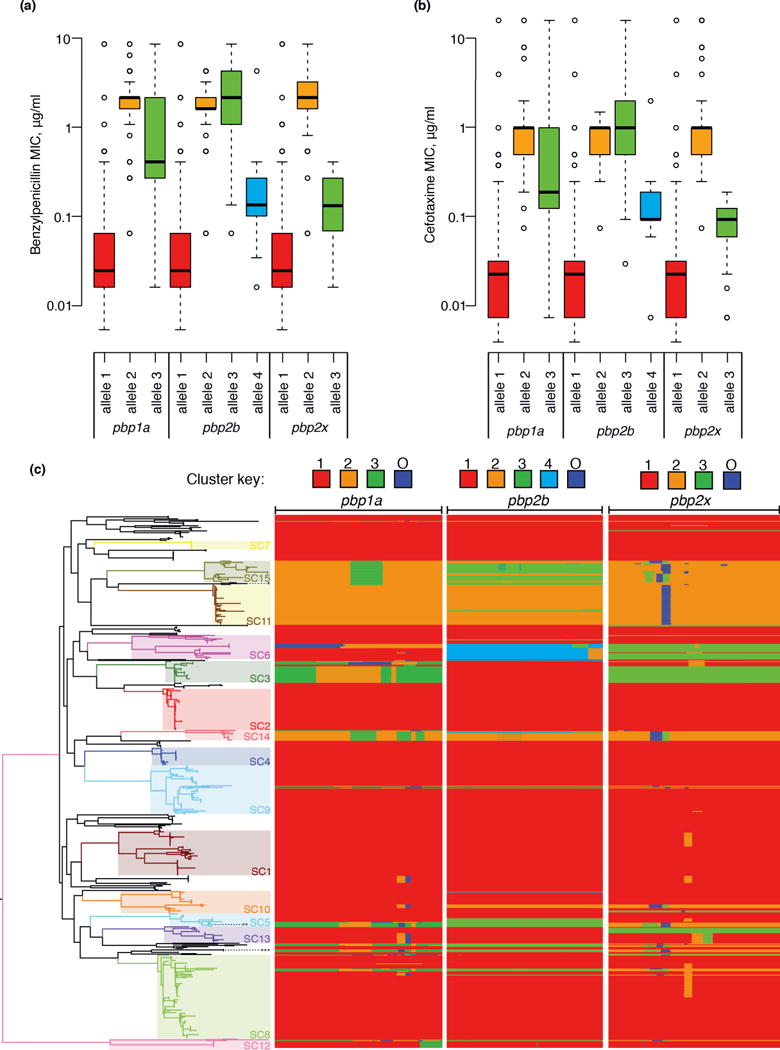
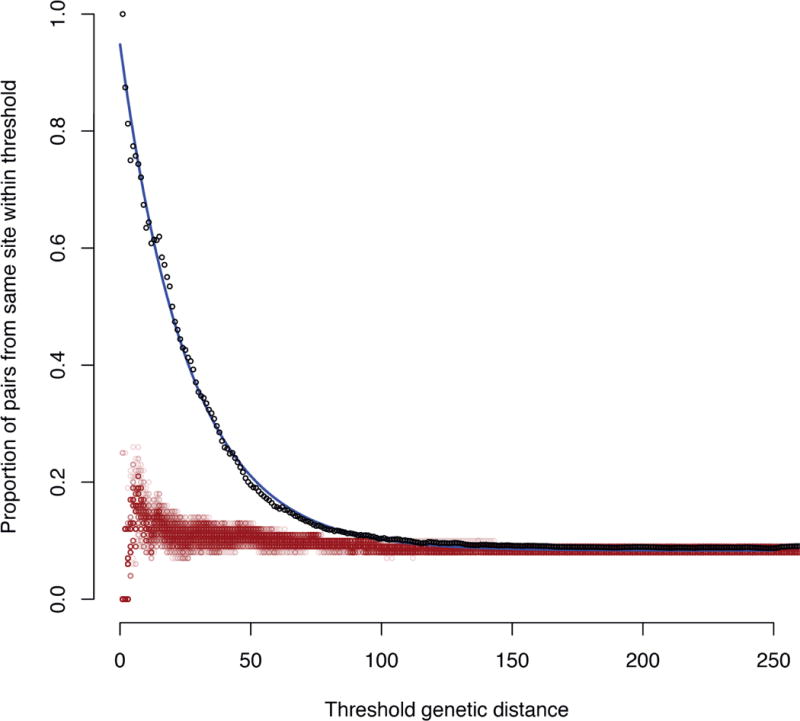
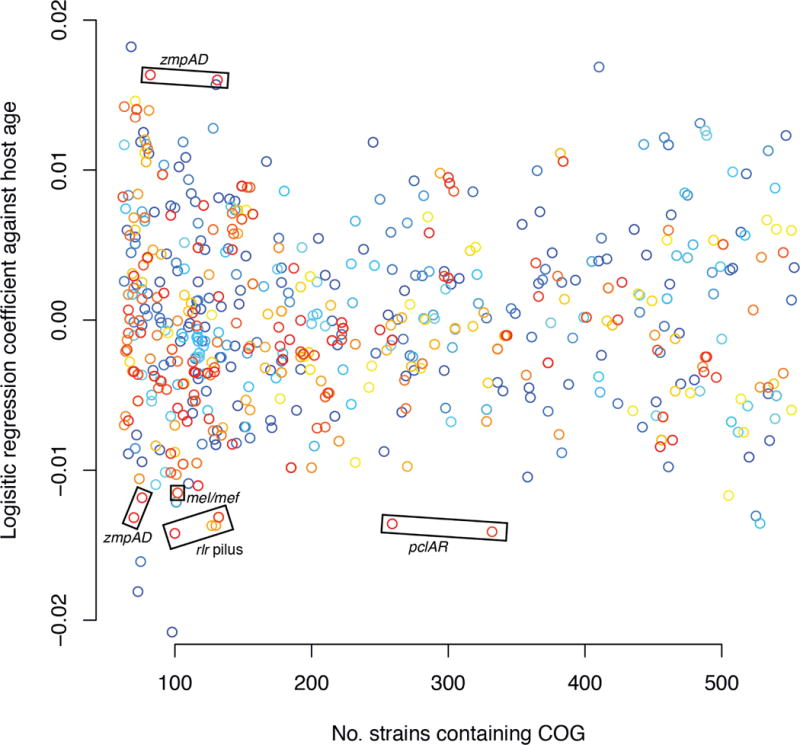
Whole-genome sequencing of 616 asymptomatically carried Streptococcus pneumoniae isolates was used to study the impact of the 7-valent pneumococcal conjugate vaccine. Comparison of closely related isolates showed the role of transformation in facilitating capsule switching to non-vaccine serotypes and the emergence of drug resistance. However, such recombination was found to occur at significantly different rates across the species, and the evolution of the population was primarily driven by changes in the frequency of distinct genotypes extant before the introduction of the vaccine. These alterations resulted in little overall effect on accessory genome composition at the population level, contrasting with the decrease in pneumococcal disease rates after the vaccine's introduction.
Figures
References
-
- Whitney CG, et al. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N Engl J Med. 2003;348:1737–46. - PubMed
-
- Steenhoff AP, Shah SS, Ratner AJ, Patil SM, McGowan KL. Emergence of vaccine-related pneumococcal serotypes as a cause of bacteremia. Clin Infect Dis. 2006;42:907–14. - PubMed
-
- Hicks LA, et al. Incidence of pneumococcal disease due to non-pneumococcal conjugate vaccine (PCV7) serotypes in the United States during the era of widespread PCV7 vaccination, 1998–2004. J Infect Dis. 2007;196:1346–54. - PubMed
-
- Pai R, et al. Postvaccine genetic structure of Streptococcus pneumoniae serotype 19A from children in the United States. J Infect Dis. 2005;192:1988–95. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
