

Exome sequencing reveals new causal mutations in children with epileptic encephalopathies
- PMID: 23647072
- PMCID: PMC3700577
- DOI: 10.1111/epi.12201
Exome sequencing reveals new causal mutations in children with epileptic encephalopathies
Abstract
Purpose: The management of epilepsy in children is particularly challenging when seizures are resistant to antiepileptic medications, or undergo many changes in seizure type over time, or have comorbid cognitive, behavioral, or motor deficits. Despite efforts to classify such epilepsies based on clinical and electroencephalographic criteria, many children never receive a definitive etiologic diagnosis. Whole exome sequencing (WES) is proving to be a highly effective method for identifying de novo variants that cause neurologic disorders, especially those associated with abnormal brain development. Herein we explore the utility of WES for identifying candidate causal de novo variants in a cohort of children with heterogeneous sporadic epilepsies without etiologic diagnoses.
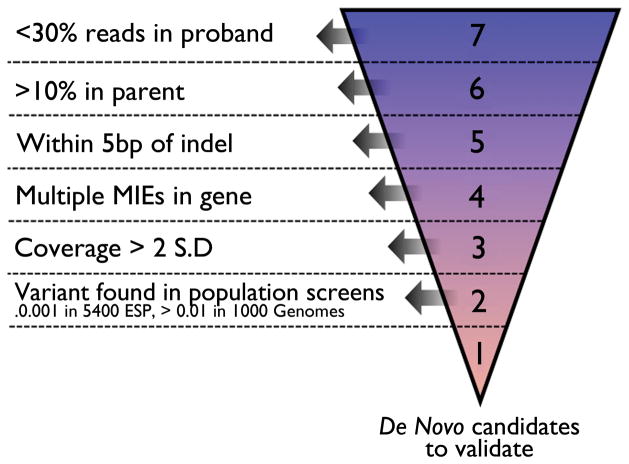
Methods: We performed WES (mean coverage approximately 40×) on 10 trios comprised of unaffected parents and a child with sporadic epilepsy characterized by difficult-to-control seizures and some combination of developmental delay, epileptic encephalopathy, autistic features, cognitive impairment, or motor deficits. Sequence processing and variant calling were performed using standard bioinformatics tools. A custom filtering system was used to prioritize de novo variants of possible functional significance for validation by Sanger sequencing.
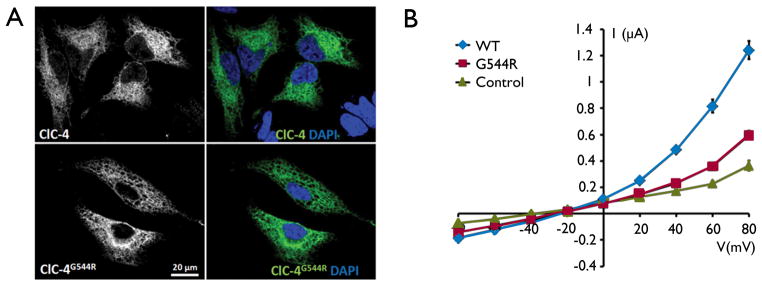
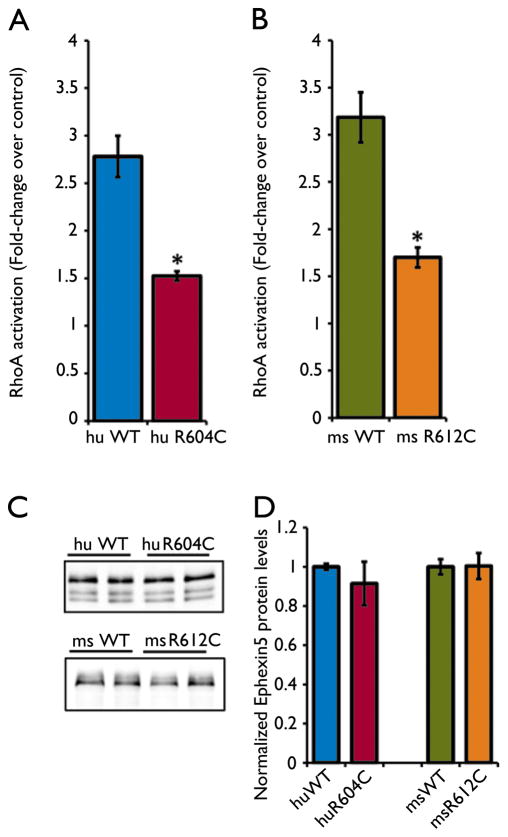
Key findings: In 9 of 10 probands, we identified one or more de novo variants predicted to alter protein function, for a total of 15. Four probands had de novo mutations in genes previously shown to harbor heterozygous mutations in patients with severe, early onset epilepsies (two in SCN1A, and one each in CDKL5 and EEF1A2). In three children, the de novo variants were in genes with functional roles that are plausibly relevant to epilepsy (KCNH5, CLCN4, and ARHGEF15). The variant in KCNH5 alters one of the highly conserved arginine residues of the voltage sensor of the encoded voltage-gated potassium channel. In vitro analyses using cell-based assays revealed that the CLCN4 mutation greatly impaired ion transport by the ClC-4 2Cl(-) /H(+) -exchanger and that the mutation in ARHGEF15 reduced GEF exchange activity of the gene product, Ephexin5, by about 50%. Of interest, these seven probands all presented with seizures within the first 6 months of life, and six of these have intractable seizures.
Significance: The finding that 7 of 10 children carried de novo mutations in genes of known or plausible clinical significance to neuronal excitability suggests that WES will be of use for the molecular genetic diagnosis of sporadic epilepsies in children, especially when seizures are of early onset and difficult to control.
Keywords: ARHGEF15; CDKL5; CLCN4; Epileptic encephalopathy; SCN1A.
Wiley Periodicals, Inc. © 2013 International League Against Epilepsy.
Conflict of interest statement
None of the authors has any conflict of interest to disclose.
Figures
References
-
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. - PubMed
-
- Berg AT, Shinnar S, Testa FM, Levy SR, Smith SN, Beckerman B. Mortality in childhood-onset epilepsy. Arch Pediatr Adolesc Med. 2004;158:1147–1152. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
