

That which does not kill me makes me stronger; combining ERK1/2 pathway inhibitors and BH3 mimetics to kill tumour cells and prevent acquired resistance
- PMID: 23647573
- PMCID: PMC3753831
- DOI: 10.1111/bph.12220
That which does not kill me makes me stronger; combining ERK1/2 pathway inhibitors and BH3 mimetics to kill tumour cells and prevent acquired resistance
Abstract
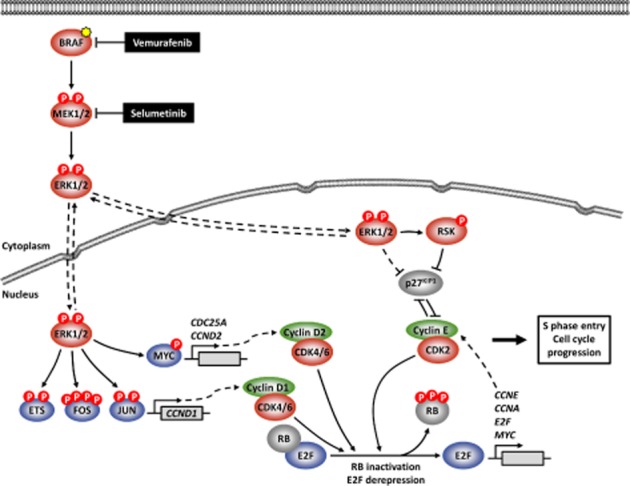
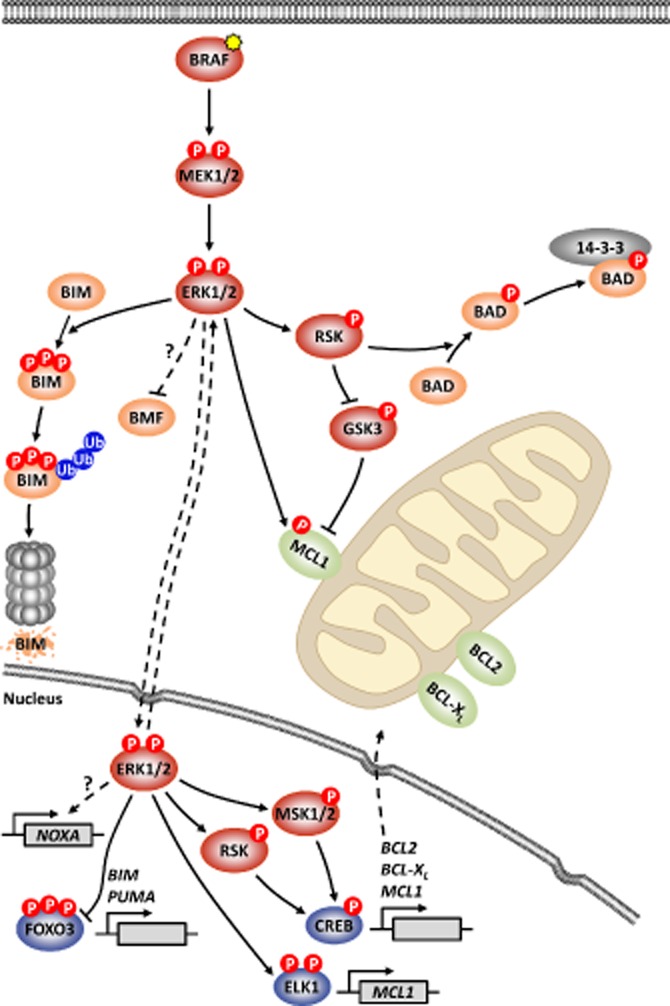
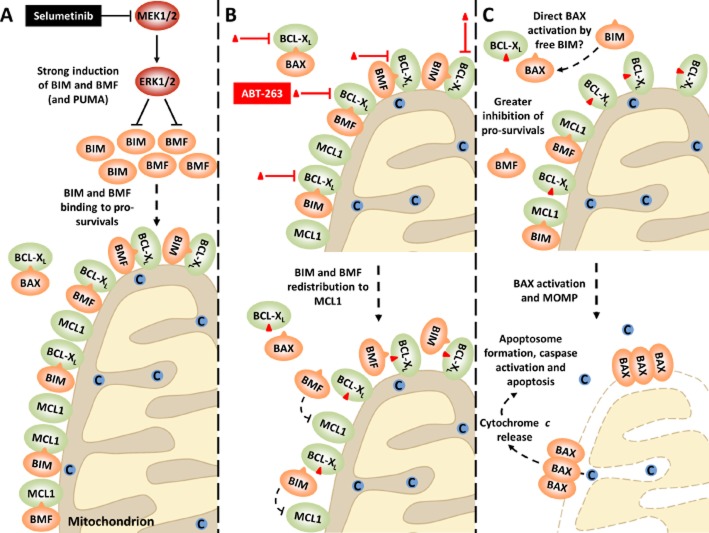
Oncogenic mutations in RAS or BRAF can drive the inappropriate activation of the ERK1/2. In many cases, tumour cells adapt to become addicted to this deregulated ERK1/2 signalling for their proliferation, providing a therapeutic window for tumour-selective growth inhibition. As a result, inhibition of ERK1/2 signalling by BRAF or MEK1/2 inhibitors is an attractive therapeutic strategy. Indeed, the first BRAF inhibitor, vemurafenib, has now been approved for clinical use, while clinical evaluation of MEK1/2 inhibitors is at an advanced stage. Despite this progress, it is apparent that tumour cells adapt quickly to these new targeted agents so that tumours with acquired resistance can emerge within 6-9 months of primary treatment. One of the major reasons for this is that tumour cells typically respond to BRAF or MEK1/2 inhibitors by undergoing a G1 cell cycle arrest rather than dying. Indeed, although inhibition of ERK1/2 invariably increases the expression of pro-apoptotic BCL2 family proteins, tumour cells undergo minimal apoptosis. This cytostatic response may simply provide the cell with the opportunity to adapt and acquire resistance. Here we discuss recent studies that demonstrate that combination of BRAF or MEK1/2 inhibitors with inhibitors of pro-survival BCL2 proteins is synthetic lethal for ERK1/2-addicted tumour cells. This combination effectively transforms the cytostatic response of BRAF and MEK1/2 inhibitors into a striking apoptotic cell death response. This not only augments the primary efficacy of BRAF and MEK1/2 inhibitors but delays the onset of acquired resistance to these agents, validating their combination in the clinic.
Linked articles: This article is part of a themed section on Emerging Therapeutic Aspects in Oncology. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.169.issue-8.
Keywords: BCL2; BRAF; ERK1/2; MEK1/2; RAS; acquired resistance; apoptosis; cancer; targeted therapeutics.
© 2013 The British Pharmacological Society.
Figures
Similar articles
-
The BH3 mimetic ABT-263 synergizes with the MEK1/2 inhibitor selumetinib/AZD6244 to promote BIM-dependent tumour cell death and inhibit acquired resistance.Biochem J. 2013 Mar 1;450(2):285-94. doi: 10.1042/BJ20121212. Biochem J. 2013. PMID: 23234544
-
Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma.J Clin Oncol. 2013 May 10;31(14):1767-74. doi: 10.1200/JCO.2012.44.7888. Epub 2013 Apr 8. J Clin Oncol. 2013. PMID: 23569304 Clinical Trial.
-
The MEK1/2 Inhibitor AZD6244 Sensitizes BRAF-Mutant Thyroid Cancer to Vemurafenib.Med Sci Monit. 2018 May 8;24:3002-3010. doi: 10.12659/MSM.910084. Med Sci Monit. 2018. Retraction in: Med Sci Monit. 2022 Mar 07;28:e936571. doi: 10.12659/MSM.936571. PMID: 29737325 Free PMC article. Retracted.
-
Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling.FEBS J. 2017 Dec;284(24):4177-4195. doi: 10.1111/febs.14122. Epub 2017 Jun 18. FEBS J. 2017. PMID: 28548464 Free PMC article. Review.
-
Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: an emerging threat to anticancer therapy.Oncogene. 2016 May 19;35(20):2547-61. doi: 10.1038/onc.2015.329. Epub 2015 Sep 14. Oncogene. 2016. PMID: 26364606 Review.
Cited by
-
Targeting melanoma's MCL1 bias unleashes the apoptotic potential of BRAF and ERK1/2 pathway inhibitors.Nat Commun. 2019 Nov 14;10(1):5167. doi: 10.1038/s41467-019-12409-w. Nat Commun. 2019. PMID: 31727888 Free PMC article.
-
SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers.Nat Commun. 2019 Jun 10;10(1):2532. doi: 10.1038/s41467-019-10367-x. Nat Commun. 2019. PMID: 31182717 Free PMC article.
-
Suppressing ERK Pathway Impairs Glycochenodeoxycholate-Mediated Survival and Drug-Resistance in Hepatocellular Carcinoma Cells.Front Oncol. 2021 Jul 13;11:663944. doi: 10.3389/fonc.2021.663944. eCollection 2021. Front Oncol. 2021. PMID: 34327135 Free PMC article.
-
Targeting BCL2-Proteins for the Treatment of Solid Tumours.Adv Med. 2014;2014:943648. doi: 10.1155/2014/943648. Epub 2014 Aug 27. Adv Med. 2014. PMID: 26556430 Free PMC article. Review.
-
Future perspectives in melanoma research: meeting report from the "Melanoma Bridge", Napoli, December 5th-8th 2013.J Transl Med. 2014 Oct 28;12:277. doi: 10.1186/s12967-014-0277-z. J Transl Med. 2014. PMID: 25348889 Free PMC article.
References
-
- Andrews M, Behren A, Chiohn F, Tebbutt N, Do H, Dobrovic A, et al. Colorectal cancer promoted in a melanoma patient receiving dabrafenib (GSK2118436) in combination with MEK1/2 inhibitor trametinib (GSK1120212) Pigment Cell Melanoma Res. 2012;25:842.
-
- Anforth RM, Blumetti TC, Kefford RF, Sharma R, Scolyer RA, Kossard S, et al. Cutaneous manifestations of dabrafenib (GSK2118436): a selective inhibitor of mutant BRAF in patients with metastatic melanoma. Br J Dermatol. 2012;167:1153–1160. - PubMed
-
- Arnault JP, Mateus C, Escudier B, Tomasic G, Wechsler J, Hollville E, et al. Skin tumors induced by sorafenib; paradoxic RAS-RAF pathway activation and oncogenic mutations of HRAS, TP53, and TGFBR1. Clin Cancer Res. 2012;18:263–272. - PubMed
-
- Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368–377. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- BBS/E/B/0000C199/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 14867/CRUK_/Cancer Research UK/United Kingdom
- BBS/E/B/000C0419/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/B/0000H248/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/E02162X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
