

Targeting unfolded protein response signaling pathways to ameliorate protein misfolding diseases
- PMID: 23647985
- PMCID: PMC5859939
- DOI: 10.1016/j.cbpa.2013.04.009
Targeting unfolded protein response signaling pathways to ameliorate protein misfolding diseases
Abstract
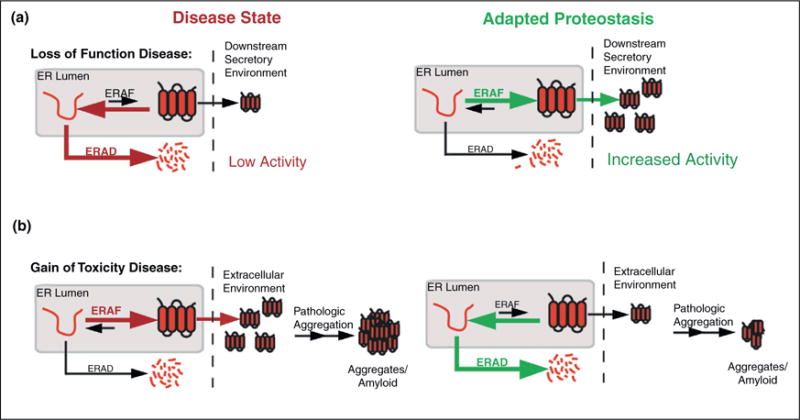
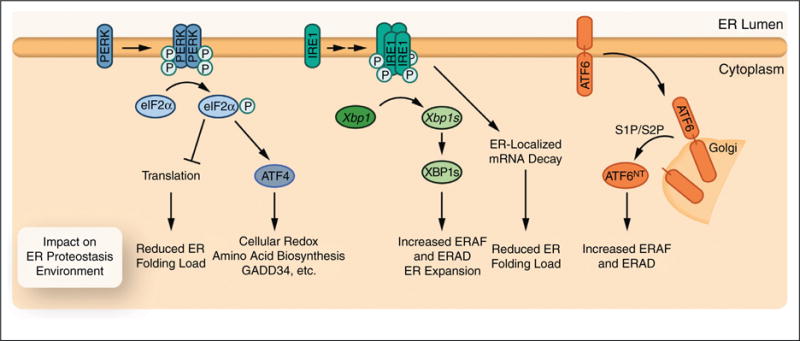
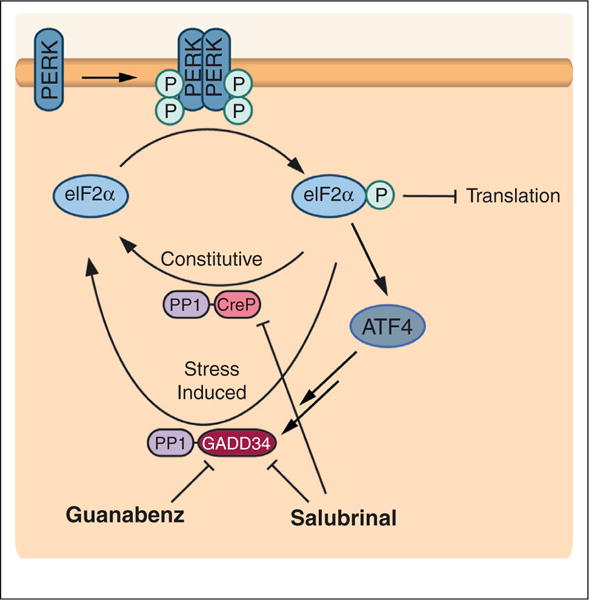
Protein homeostasis (or proteostasis) within the endoplasmic reticulum (ER) is regulated by the unfolded protein response (UPR). The UPR consists of three integrated signaling pathways activated by the accumulation of misfolded proteins within the ER lumen. Activation of the UPR alters ER proteostasis through translational attenuation of new protein synthesis and transcriptional remodeling of ER proteostasis pathways, providing a mechanism to adapt ER proteostasis in response to cellular stress. The capacity of the UPR to alter ER proteostasis suggests that exogenous manipulation of UPR signaling pathways offers therapeutic promise to alter the fate of pathologic proteins associated with human protein misfolding diseases. Here, we discuss the therapeutic potential of exogenous UPR activation to treat human disease and highlight specific small molecule approaches for regulating UPR signaling that could be beneficial to treat protein misfolding diseases.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Small molecule strategies to harness the unfolded protein response: where do we go from here?J Biol Chem. 2020 Nov 13;295(46):15692-15711. doi: 10.1074/jbc.REV120.010218. Epub 2020 Sep 4. J Biol Chem. 2020. PMID: 32887796 Free PMC article. Review.
-
Underlying mechanisms and chemical/biochemical therapeutic approaches to ameliorate protein misfolding neurodegenerative diseases.Biofactors. 2017 Nov;43(6):737-759. doi: 10.1002/biof.1264. Epub 2016 Feb 22. Biofactors. 2017. PMID: 26899445 Review.
-
Endoplasmic reticulum proteostasis: a key checkpoint in cancer.Am J Physiol Cell Physiol. 2017 Feb 1;312(2):C93-C102. doi: 10.1152/ajpcell.00266.2016. Epub 2016 Nov 16. Am J Physiol Cell Physiol. 2017. PMID: 27856431 Free PMC article. Review.
-
The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis.Int J Biochem Cell Biol. 2015 Apr;61:45-52. doi: 10.1016/j.biocel.2015.01.015. Epub 2015 Feb 7. Int J Biochem Cell Biol. 2015. PMID: 25660369 Review.
-
Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo.Proc Natl Acad Sci U S A. 2014 Jun 3;111(22):E2271-80. doi: 10.1073/pnas.1318262111. Epub 2014 May 19. Proc Natl Acad Sci U S A. 2014. PMID: 24843123 Free PMC article.
Cited by
-
Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch.Elife. 2016 Jul 20;5:e11878. doi: 10.7554/eLife.11878. Elife. 2016. PMID: 27435960 Free PMC article.
-
The endoplasmic reticulum proteostasis network profoundly shapes the protein sequence space accessible to HIV envelope.PLoS Biol. 2022 Feb 18;20(2):e3001569. doi: 10.1371/journal.pbio.3001569. eCollection 2022 Feb. PLoS Biol. 2022. PMID: 35180219 Free PMC article.
-
Protein Aggregation and Disaggregation in Cells and Development.J Mol Biol. 2021 Oct 15;433(21):167215. doi: 10.1016/j.jmb.2021.167215. Epub 2021 Aug 24. J Mol Biol. 2021. PMID: 34450138 Free PMC article. Review.
-
Hallmarks of therapeutic management of the cystic fibrosis functional landscape.J Cyst Fibros. 2015 Nov;14(6):687-99. doi: 10.1016/j.jcf.2015.09.006. Epub 2015 Oct 29. J Cyst Fibros. 2015. PMID: 26526359 Free PMC article. Review.
-
Research hotspots and frontiers of endoplasmic reticulum in glomerular podocytes: a bibliometric and visual analysis from 2005 to 2023.Front Pharmacol. 2025 Jan 7;15:1488340. doi: 10.3389/fphar.2024.1488340. eCollection 2024. Front Pharmacol. 2025. PMID: 39840101 Free PMC article.
References
-
- Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. - PubMed
-
- Wiseman RL, et al. An adaptable standard for protein export from the endoplasmic reticulum. Cell. 2007;131:809–821. - PubMed
-
- Powers ET, et al. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. - PubMed
-
- Balch WE, et al. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
