

Reconstituting protein interaction networks using parameter-dependent domain-domain interactions
- PMID: 23651452
- PMCID: PMC3660195
- DOI: 10.1186/1471-2105-14-154
Reconstituting protein interaction networks using parameter-dependent domain-domain interactions
Abstract
Background: We can describe protein-protein interactions (PPIs) as sets of distinct domain-domain interactions (DDIs) that mediate the physical interactions between proteins. Experimental data confirm that DDIs are more consistent than their corresponding PPIs, lending support to the notion that analyses of DDIs may improve our understanding of PPIs and lead to further insights into cellular function, disease, and evolution. However, currently available experimental DDI data cover only a small fraction of all existing PPIs and, in the absence of structural data, determining which particular DDI mediates any given PPI is a challenge.
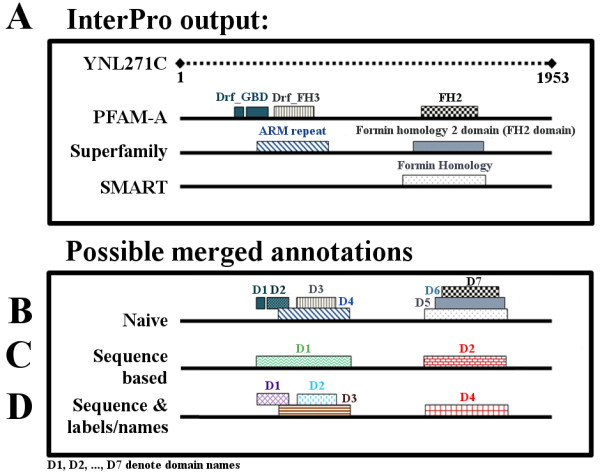
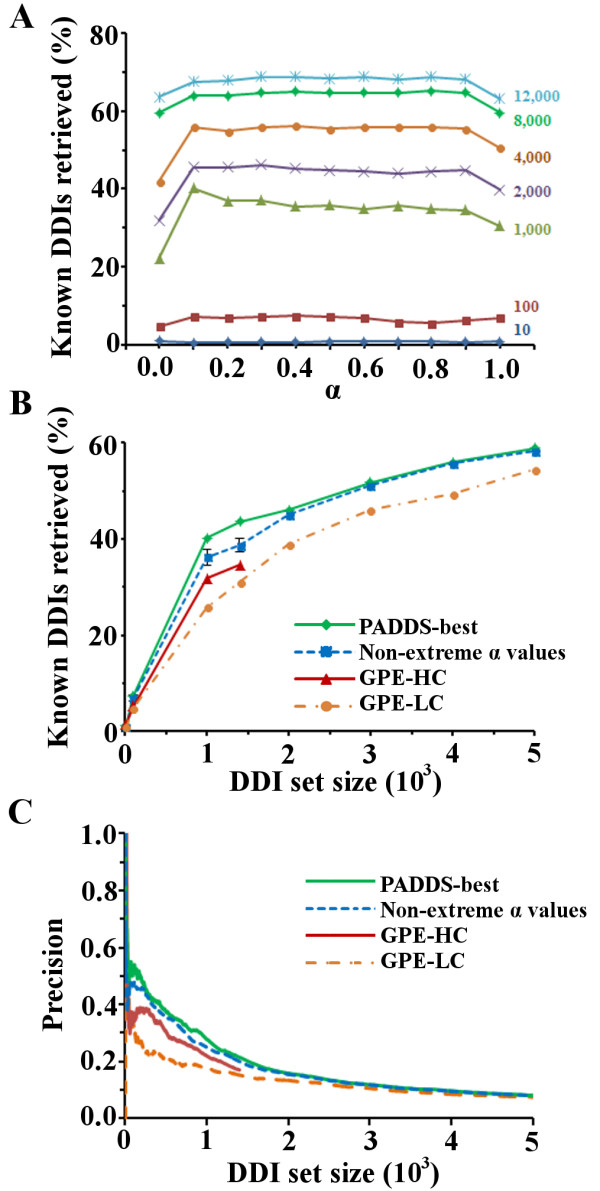
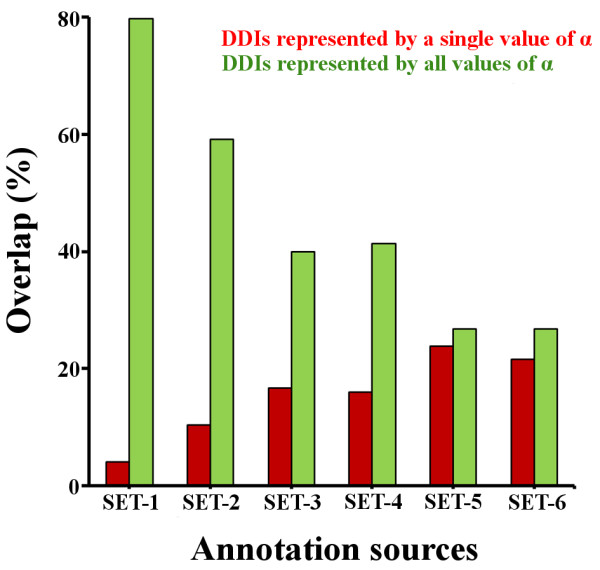
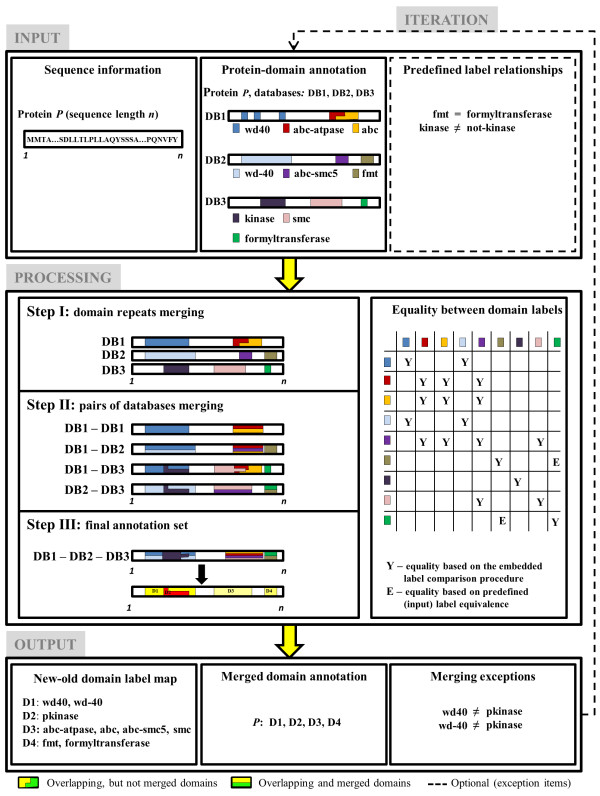
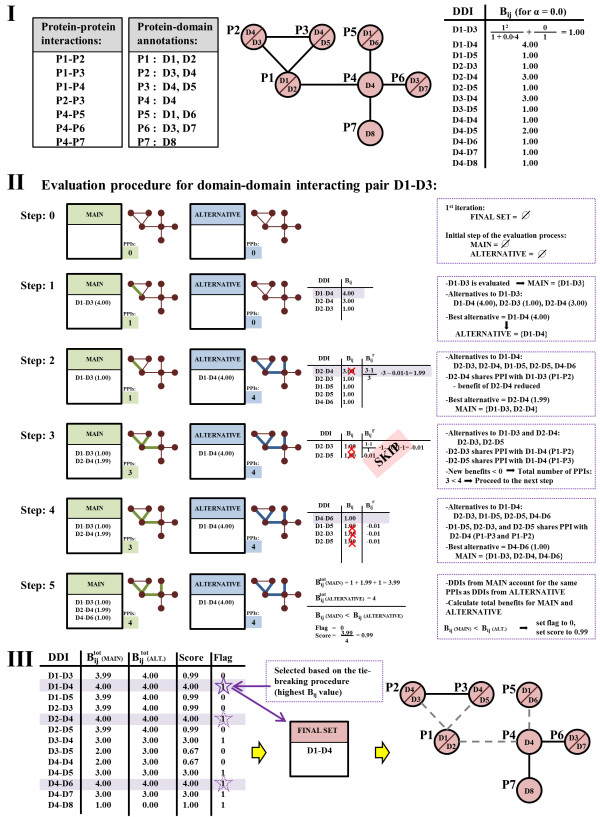
Results: We present two contributions to the field of domain interaction analysis. First, we introduce a novel computational strategy to merge domain annotation data from multiple databases. We show that when we merged yeast domain annotations from six annotation databases we increased the average number of domains per protein from 1.05 to 2.44, bringing it closer to the estimated average value of 3. Second, we introduce a novel computational method, parameter-dependent DDI selection (PADDS), which, given a set of PPIs, extracts a small set of domain pairs that can reconstruct the original set of protein interactions, while attempting to minimize false positives. Based on a set of PPIs from multiple organisms, our method extracted 27% more experimentally detected DDIs than existing computational approaches.
Conclusions: We have provided a method to merge domain annotation data from multiple sources, ensuring large and consistent domain annotation for any given organism. Moreover, we provided a method to extract a small set of DDIs from the underlying set of PPIs and we showed that, in contrast to existing approaches, our method was not biased towards DDIs with low or high occurrence counts. Finally, we used these two methods to highlight the influence of the underlying annotation density on the characteristics of extracted DDIs. Although increased annotations greatly expanded the possible DDIs, the lack of knowledge of the true biological false positive interactions still prevents an unambiguous assignment of domain interactions responsible for all protein network interactions.Executable files and examples are given at: http://www.bhsai.org/downloads/padds/
Figures
References
-
- von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P. Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 2002;417(6887):399–403. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
