

Resistance to BH3 mimetic S1 in SCLC cells that up-regulate and phosphorylate Bcl-2 through ERK1/2
- PMID: 23651505
- PMCID: PMC3724116
- DOI: 10.1111/bph.12243
Resistance to BH3 mimetic S1 in SCLC cells that up-regulate and phosphorylate Bcl-2 through ERK1/2
Abstract
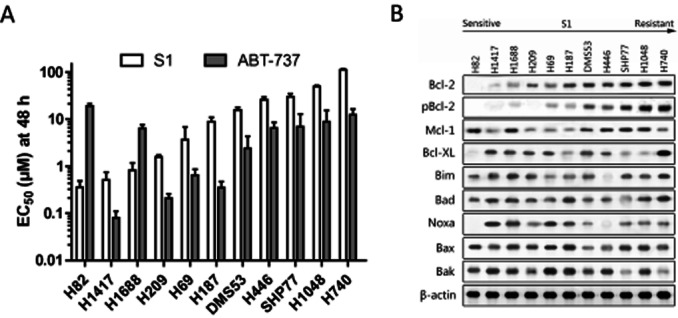
Background and purpose: B cell lymphoma 2 (Bcl-2) is a central regulator of cell survival that is overexpressed in the majority of small-cell lung cancers (SCLC) and contributes to both malignant transformation and therapeutic resistance. The purpose of this work was to study the key factors that determine the sensitivity of SCLC cells to Bcl-2 homology domain-3 (BH3) mimetic S1 and the mechanism underlying the resistance of BH3 mimetics.
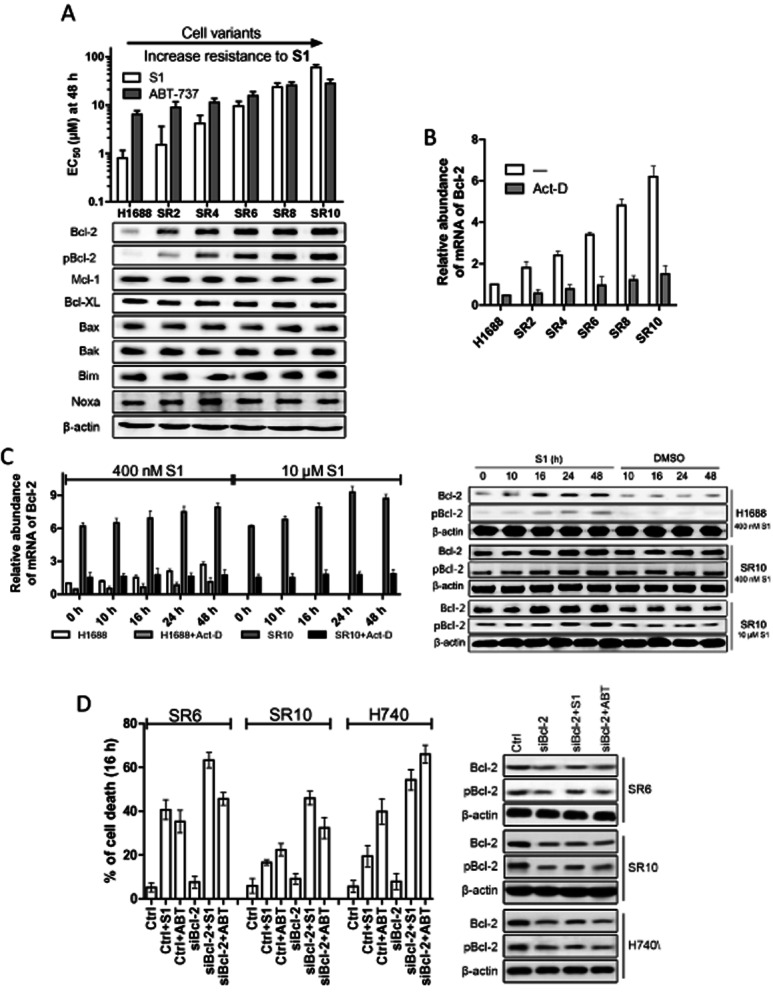
Experimental approaches: Western blot was used to evaluate the contribution of Bcl-2 family members to the cellular response of SCLC cell lines to S1. Acquired resistant cells were derived from initially sensitive H1688 cells. Quantitative PCR and gene silencing were performed to investigate Bcl-2 up-regulation.
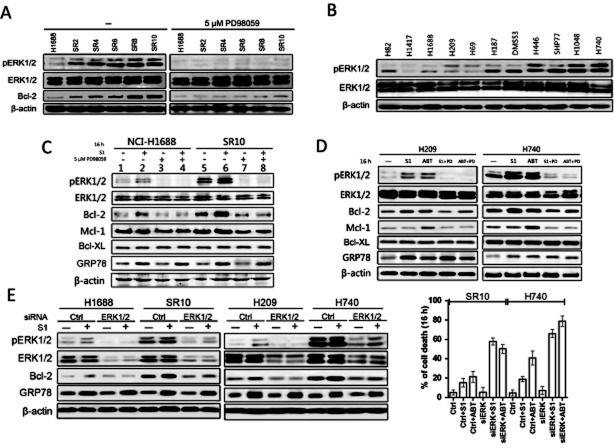
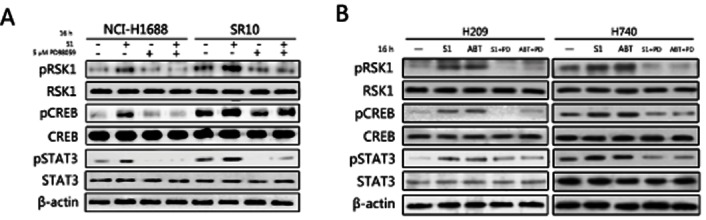
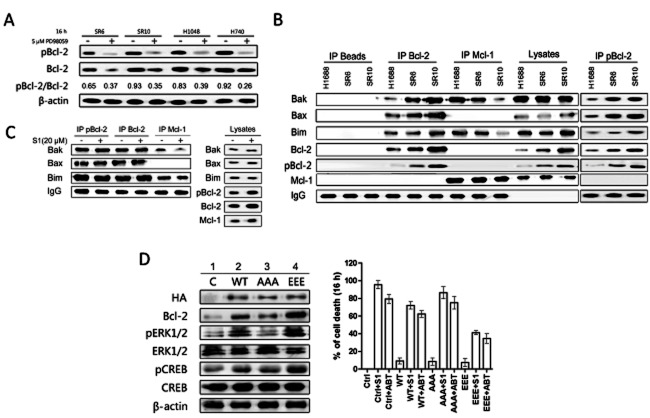
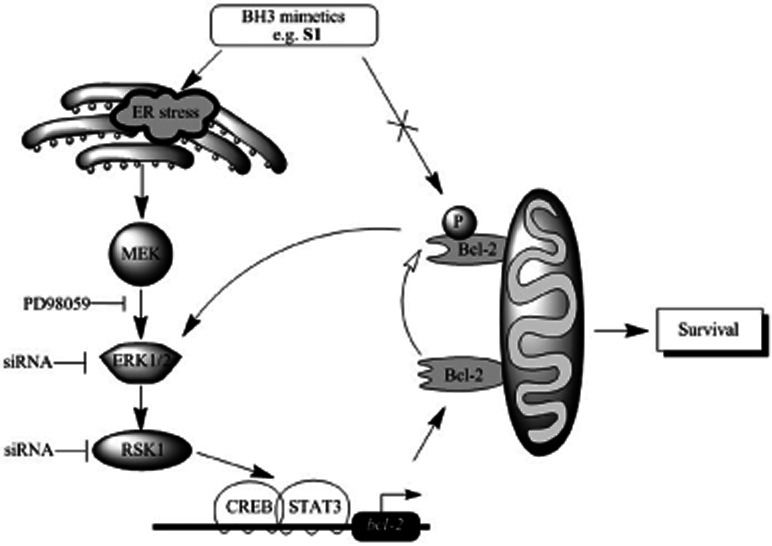
Key results: A progressive increase in the relative levels of Bcl-2 and phosphorylated Bcl-2 (pBcl-2) characterized the increased de novo and acquired resistance of SCLC cell lines. Furthermore, acute treatment of S1 induced Bcl-2 expression and phosphorylation. We showed that BH3 mimetics, including S1 and ABT-737, induced endoplasmic reticulum (ER) stress and then activated MAPK/ERK pathway. The dual function of MAPK/ERK pathway in defining BH3 mimetics was illustrated; ERK1/2 activation leaded to Bcl-2 transcriptional up-regulation and sustained phosphorylation in naïve and acquired resistant SCLC cells. pBcl-2 played a key role in creating resistance of S1 and ABT-737 not only by sequestrating pro-apoptotic proteins, but also sequestrating a positive feedback to promote ERK1/2 activation.
Conclusions and implications: These results provide significant novel insights into the molecular mechanisms for crosstalk between ER stress and endogenously apoptotic pathways in SCLC following BH3 mimetics treatment.
Keywords: BH3 mimetics; Bcl-2 phosphorylation; ERK1/2; SCLC.
© 2013 The British Pharmacological Society.
Figures
References
-
- Breton C, Story MD, Meyn RE. Bcl-2 expression correlates with apoptosis induction but not loss of clonogenic survival in small cell lung cancer cell lines treated with etoposide. Anticancer Drugs. 1998;9:751–757. - PubMed
-
- Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Ann Rev Immunol. 1998;16:395–419. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
