

Klotho and chronic kidney disease
- PMID: 23652549
- PMCID: PMC3911771
- DOI: 10.1159/000346778
Klotho and chronic kidney disease
Abstract
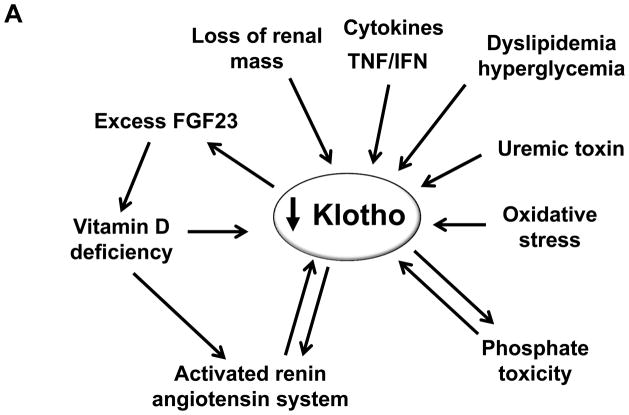
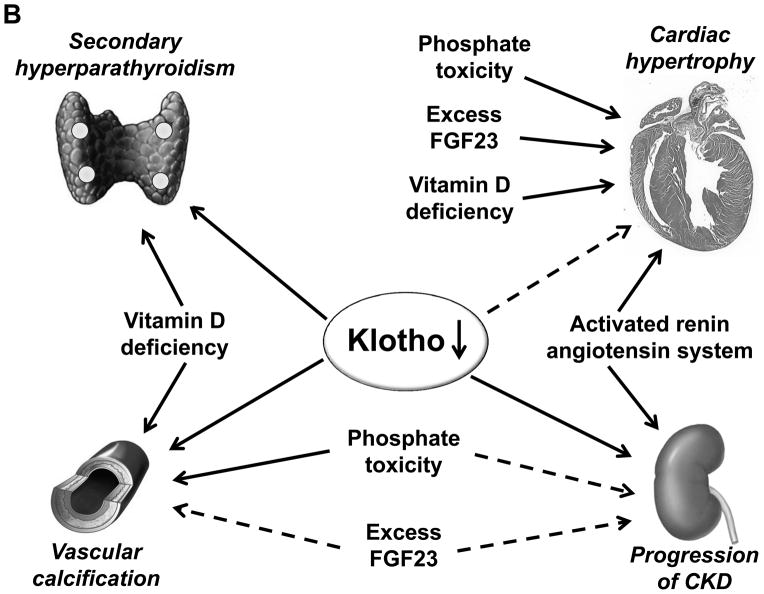
Through alternative splicing, Klotho protein exists both as a secreted and a membrane form whose extracellular domain could be shed from the cell surface by secretases and released into the circulation to act as endocrine factor. Unlike membrane Klotho which functions as a coreceptor for fibroblast growth factor-23 (FGF23) to modulate FGF23 signal transduction, soluble Klotho is a multifunction protein present in the biological fluids including blood, urine and cerebrospinal fluid and plays important roles in antiaging, energy metabolism, inhibition of Wnt signaling, antioxidation, modulation of ion transport, control of parathyroid hormone and 1,25(OH)2VD3 production, and antagonism of renin-angiotensin-aldosterone system. Emerging evidence from clinical and basic studies reveal that chronic kidney disease is a state of endocrine and renal Klotho deficiency, which may serve as an early biomarker and a pathogenic contributor to chronic progression and complications in chronic kidney disease including vascular calcification, cardiac hypertrophy, and secondary hyperparathyroidism. Supplementation of exogenous Klotho and/or upregulation of endogenous Klotho production by using rennin angiotensin system inhibitors, HMG CoA reductase inhibitors, vitamin D analogues, peroxisome proliferator-activated receptors-gamma agonists, or anti-oxidants may confer renoprotection from oxidation and suppression of renal fibrosis, and also on prevention or alleviation of complications in chronic kidney disease. Therefore, Klotho is a highly promising candidate on the horizon as an early biomarker, and as a novel therapeutic agent for chronic kidney disease.
Copyright © 2013 S. Karger AG, Basel.
Figures

References
-
- Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, Van Lente F, Levey AS. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–2047. - PubMed
-
- Levey AS, Atkins R, Coresh J, Cohen EP, Collins AJ, Eckardt KU, Nahas ME, Jaber BL, Jadoul M, Levin A, Powe NR, Rossert J, Wheeler DC, Lameire N, Eknoyan G. Chronic kidney disease as a global public health problem: approaches and initiatives - a position statement from Kidney Disease Improving Global Outcomes. Kidney Int. 2007;72:247–259. - PubMed
-
- Takeshita K, Fujimori T, Kurotaki Y, Honjo H, Tsujikawa H, Yasui K, Lee JK, Kamiya K, Kitaichi K, Yamamoto K, Ito M, Kondo T, Iino S, Inden Y, Hirai M, Murohara T, Kodama I, Nabeshima Y. Sinoatrial node dysfunction and early unexpected death of mice with a defect of klotho gene expression. Circulation. 2004;109:1776–1782. - PubMed
-
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
