

Ataxin-3 and its e3 partners: implications for machado-joseph disease
- PMID: 23653622
- PMCID: PMC3644722
- DOI: 10.3389/fneur.2013.00046
Ataxin-3 and its e3 partners: implications for machado-joseph disease
Abstract
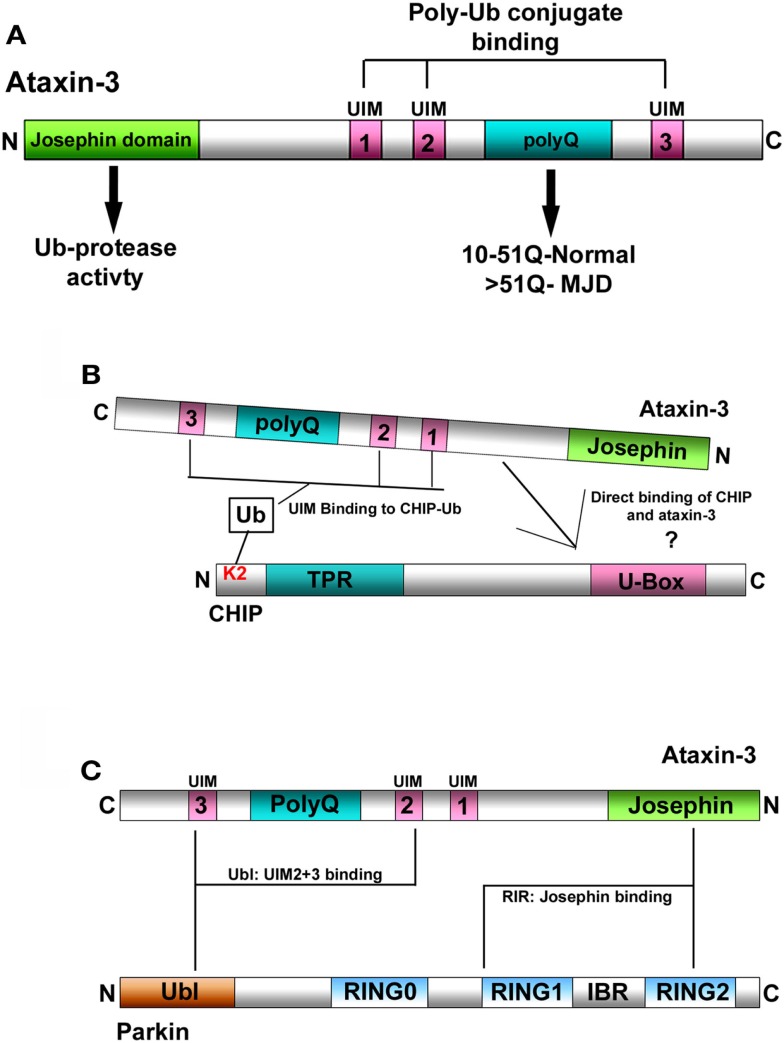
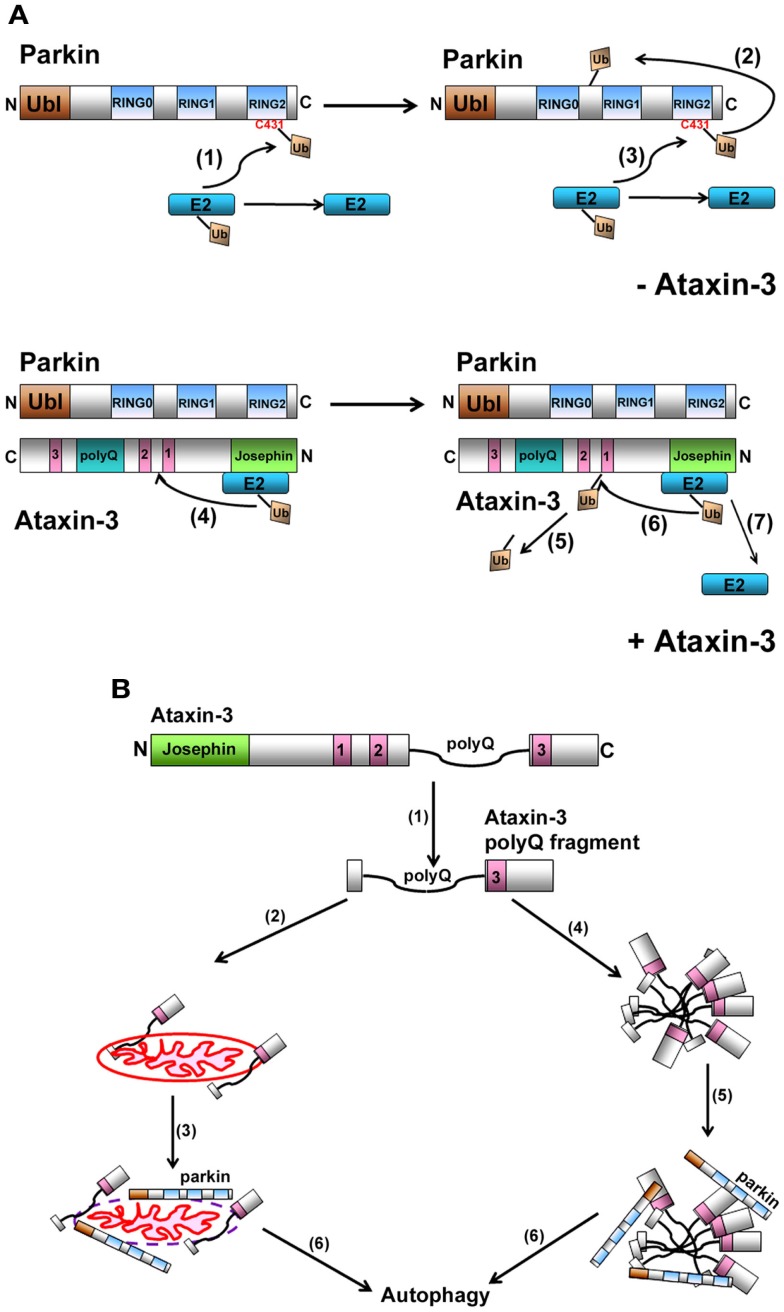
Machado-Joseph disease (MJD) is the most common dominant inherited ataxia worldwide, caused by an unstable CAG trinucleotide expansion mutation within the SCA3 gene resulting in an expanded polyglutamine tract within the ataxin-3 protein. Ataxin-3 functions as a deubiquitinating enzyme (DUB), within the Ub system and whilst many DUBs are known to partner with and deubiquitinate specific E3-Ub ligases, ataxin-3 had no identified E3 partner until recent studies implicated parkin and CHIP, two neuroprotective E3 ligases. MJD often presents with symptoms of Parkinson disease (PD), which led to identification of parkin as a novel E3-Ub ligase whose activity was regulated by ataxin-3-mediated deubiquitination. Findings from these studies also revealed an unexpected convergence upon the E2-Ub-conjugating enzyme in the regulation of an E3/DUBenzyme pair. Moreover, mutant but not wild-type ataxin-3 promotes the clearance of parkin via the autophagy pathway, raising the intriguing possibility that increased turnover of parkin may contribute to the pathogenesis of MJD and help explain some of the Parkinsonian features in MJD. In addition to parkin, the U-box E3 ligase CHIP, a neuroprotective E3 implicated in protein quality control, was identified as a second E3 partner of ataxin-3, with ataxin-3 regulating the ability of CHIP to ubiquitinate itself. Indeed, ataxin-3 not only deubiquitinated CHIP, but also trimmed Ub conjugates on CHIP substrates, thereby regulating the length of Ub chains. Interestingly, when expanded ataxin-3 was present, CHIP levels were also reduced in the brains of MJD transgenic mice, raising the possibility that loss of one or both E3 partners may be a contributing factor in the pathogenesis of SCA3. In this review we discuss the implications from these studies and describe the importance of these findings in helping us understand the molecular processes involved in SCA3 and other neurodegenerative disorders.
Keywords: CHIP; Machado–Joseph disease; Parkinson’s disease; ataxin-3; parkin; polyglutamine expansion.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources