

Perspective: Stochastic algorithms for chemical kinetics
- PMID: 23656106
- PMCID: PMC3656953
- DOI: 10.1063/1.4801941
Perspective: Stochastic algorithms for chemical kinetics
Abstract
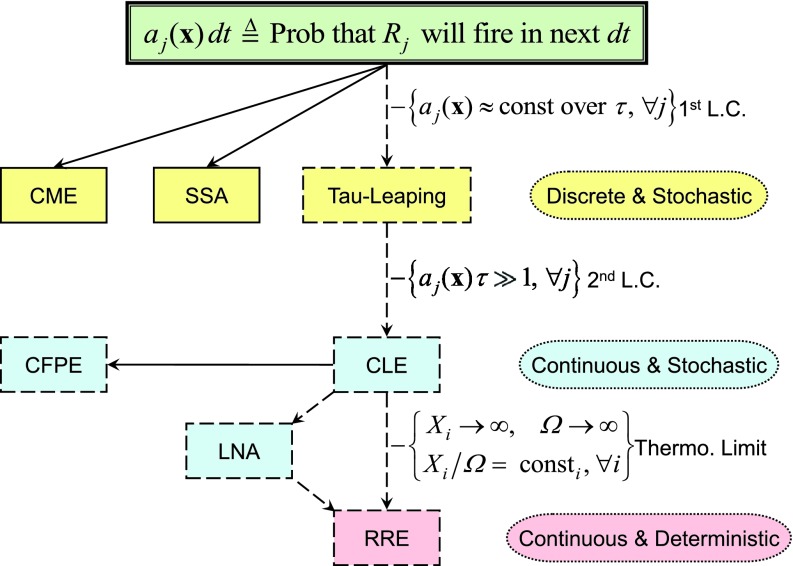
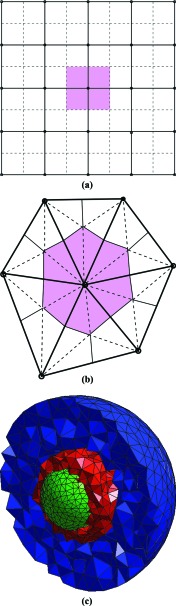
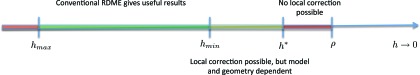
We outline our perspective on stochastic chemical kinetics, paying particular attention to numerical simulation algorithms. We first focus on dilute, well-mixed systems, whose description using ordinary differential equations has served as the basis for traditional chemical kinetics for the past 150 years. For such systems, we review the physical and mathematical rationale for a discrete-stochastic approach, and for the approximations that need to be made in order to regain the traditional continuous-deterministic description. We next take note of some of the more promising strategies for dealing stochastically with stiff systems, rare events, and sensitivity analysis. Finally, we review some recent efforts to adapt and extend the discrete-stochastic approach to systems that are not well-mixed. In that currently developing area, we focus mainly on the strategy of subdividing the system into well-mixed subvolumes, and then simulating diffusional transfers of reactant molecules between adjacent subvolumes together with chemical reactions inside the subvolumes.
Figures
Similar articles
-
Stochastic simulation of chemical kinetics.Annu Rev Phys Chem. 2007;58:35-55. doi: 10.1146/annurev.physchem.58.032806.104637. Annu Rev Phys Chem. 2007. PMID: 17037977 Review.
-
The multinomial simulation algorithm for discrete stochastic simulation of reaction-diffusion systems.J Chem Phys. 2009 Mar 7;130(9):094104. doi: 10.1063/1.3074302. J Chem Phys. 2009. PMID: 19275393 Free PMC article.
-
An accelerated algorithm for discrete stochastic simulation of reaction-diffusion systems using gradient-based diffusion and tau-leaping.J Chem Phys. 2011 Apr 21;134(15):154103. doi: 10.1063/1.3572335. J Chem Phys. 2011. PMID: 21513371 Free PMC article.
-
On the origins of approximations for stochastic chemical kinetics.J Chem Phys. 2005 Oct 22;123(16):164115. doi: 10.1063/1.2062048. J Chem Phys. 2005. PMID: 16268689
-
Unified representation of Life's basic properties by a 3-species Stochastic Cubic Autocatalytic Reaction-Diffusion system of equations.Phys Life Rev. 2022 Jul;41:64-83. doi: 10.1016/j.plrev.2022.03.003. Epub 2022 May 13. Phys Life Rev. 2022. PMID: 35594602 Review.
Cited by
-
Incorporating domain growth into hybrid methods for reaction-diffusion systems.J R Soc Interface. 2021 Apr;18(177):20201047. doi: 10.1098/rsif.2020.1047. Epub 2021 Apr 14. J R Soc Interface. 2021. PMID: 33849339 Free PMC article.
-
A multiscale compartment-based model of stochastic gene regulatory networks using hitting-time analysis.J Chem Phys. 2021 May 14;154(18):184105. doi: 10.1063/5.0010764. J Chem Phys. 2021. PMID: 34241042 Free PMC article.
-
Close agreement between deterministic versus stochastic modeling of first-passage time to vesicle fusion.Biophys J. 2022 Dec 6;121(23):4569-4584. doi: 10.1016/j.bpj.2022.10.033. Epub 2022 Oct 29. Biophys J. 2022. PMID: 36815708 Free PMC article.
-
Scalable and flexible inference framework for stochastic dynamic single-cell models.PLoS Comput Biol. 2022 May 19;18(5):e1010082. doi: 10.1371/journal.pcbi.1010082. eCollection 2022 May. PLoS Comput Biol. 2022. PMID: 35588132 Free PMC article.
-
GillesPy2: A Biochemical Modeling Framework for Simulation Driven Biological Discovery.Lett Biomath. 2023 Jan 10;10(1):87-103. Epub 2023 May 26. Lett Biomath. 2023. PMID: 37655179 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous