

Proteolytic activation transforms heparin cofactor II into a host defense molecule
- PMID: 23656734
- PMCID: PMC3677170
- DOI: 10.4049/jimmunol.1203030
Proteolytic activation transforms heparin cofactor II into a host defense molecule
Abstract
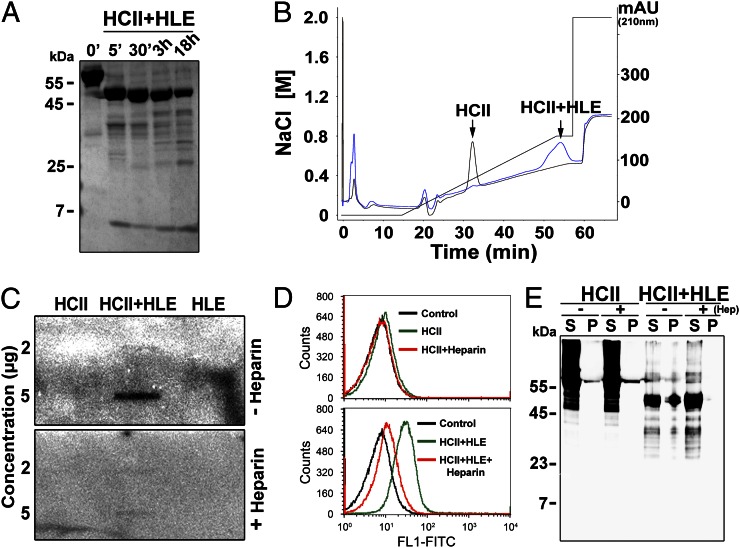
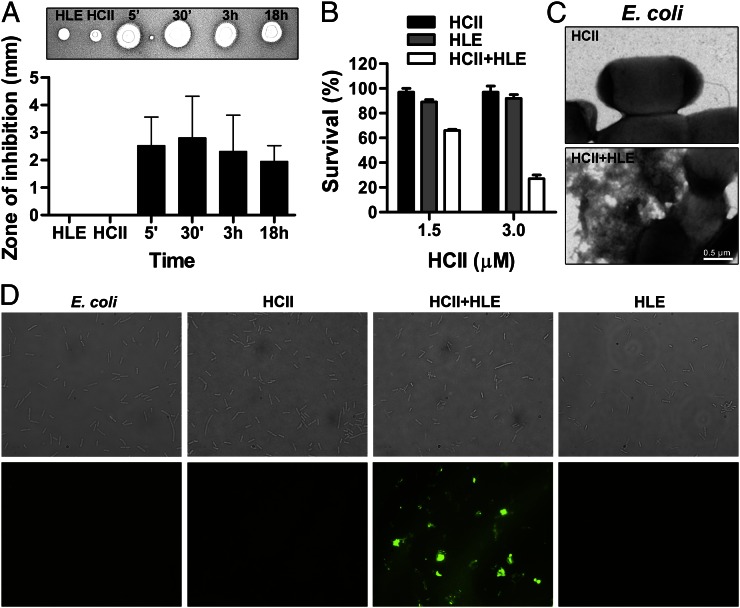
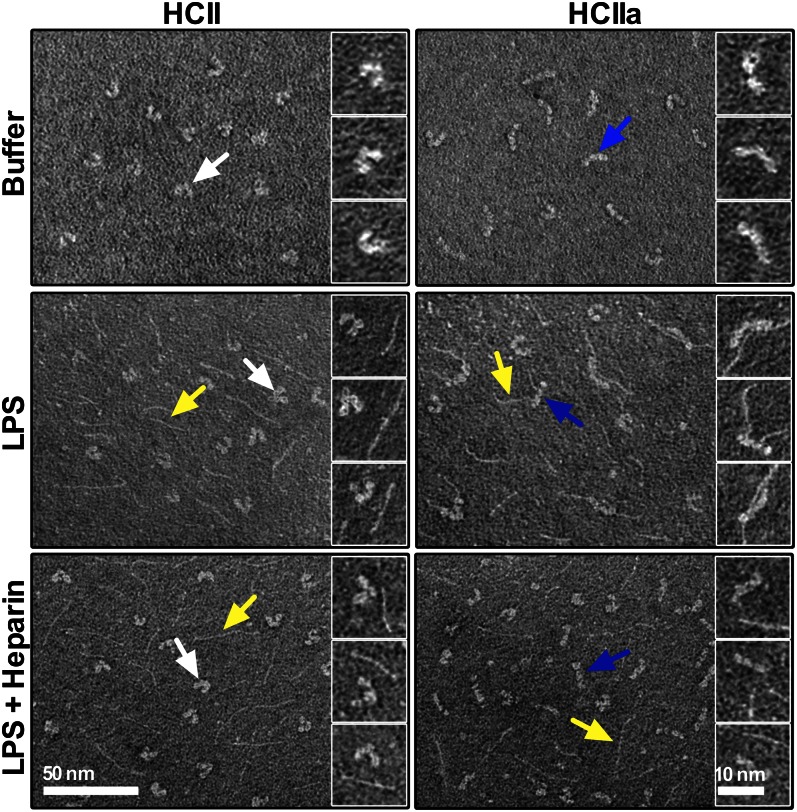
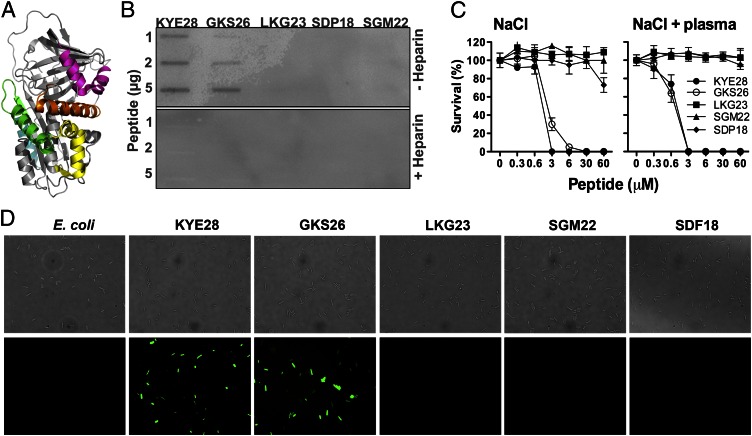
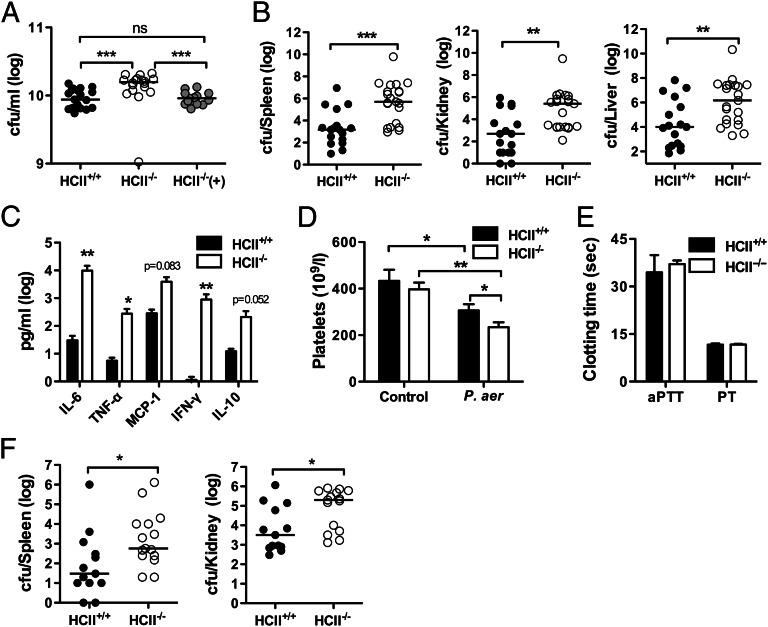
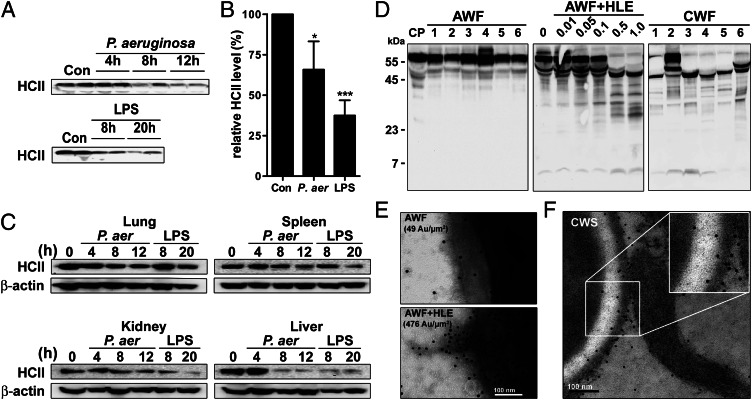
The abundant serine proteinase inhibitor heparin cofactor II (HCII) has been proposed to inhibit extravascular thrombin. However, the exact physiological role of this plasma protein remains enigmatic. In this study, we demonstrate a previously unknown role for HCII in host defense. Proteolytic cleavage of the molecule induced a conformational change, thereby inducing endotoxin-binding and antimicrobial properties. Analyses employing representative peptide epitopes mapped these effects to helices A and D. Mice deficient in HCII showed increased susceptibility to invasive infection by Pseudomonas aeruginosa, along with a significantly increased cytokine response. Correspondingly, decreased levels of HCII were observed in wild-type animals challenged with bacteria or endotoxin. In humans, proteolytically cleaved HCII forms were detected during wounding and in association with bacteria. Thus, the protease-induced uncovering of cryptic epitopes in HCII, which transforms the molecule into a host defense factor, represents a previously unknown regulatory mechanism in HCII biology and innate immunity.
Figures
References
-
- Huntington J. A. 2006. Shape-shifting serpins—advantages of a mobile mechanism. Trends Biochem. Sci. 31: 427–435 - PubMed
-
- Villa P., Aznar J., Vaya A., España F., Ferrando F., Mira Y., Estellés A. 1999. Hereditary homozygous heparin cofactor II deficiency and the risk of developing venous thrombosis. Thromb. Haemost. 82: 1011–1014 - PubMed
-
- Tollefsen D. M. 2010. Vascular dermatan sulfate and heparin cofactor II. Prog Mol Biol Transl Sci 93: 351–372 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
