

Mechanism of IL-1β modulation of intestinal epithelial barrier involves p38 kinase and activating transcription factor-2 activation
- PMID: 23656735
- PMCID: PMC3677168
- DOI: 10.4049/jimmunol.1201876
Mechanism of IL-1β modulation of intestinal epithelial barrier involves p38 kinase and activating transcription factor-2 activation
Abstract
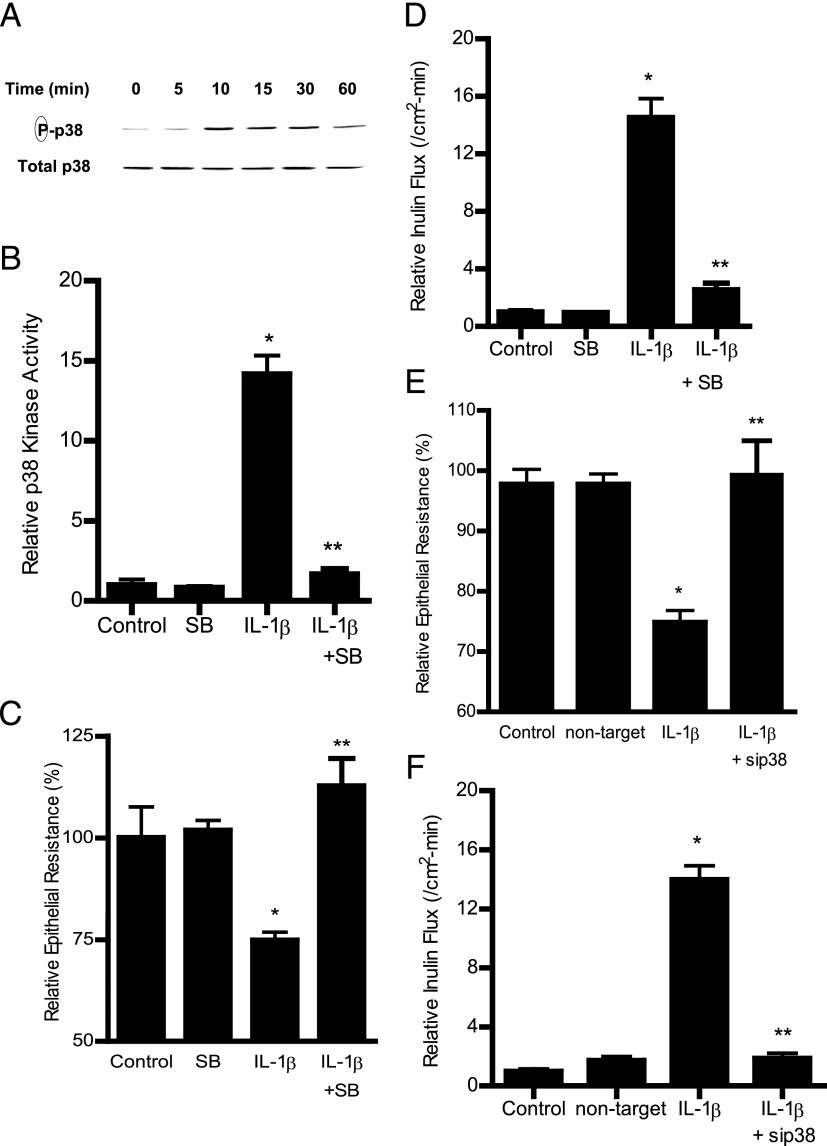
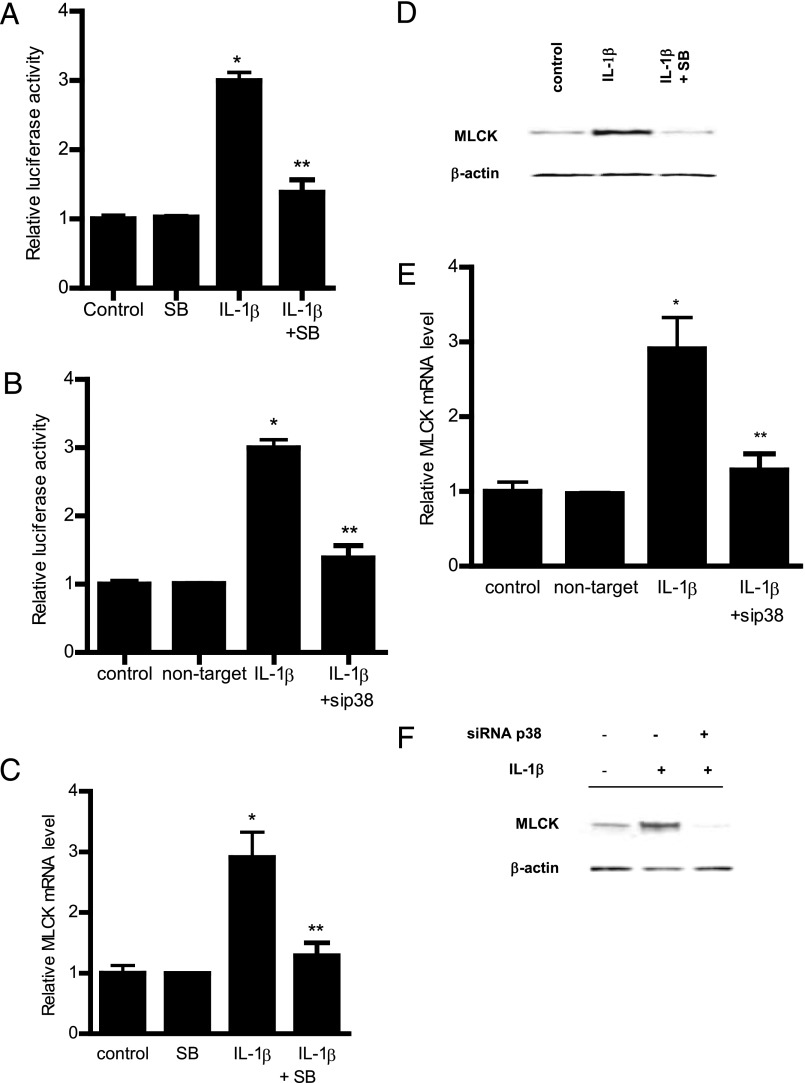
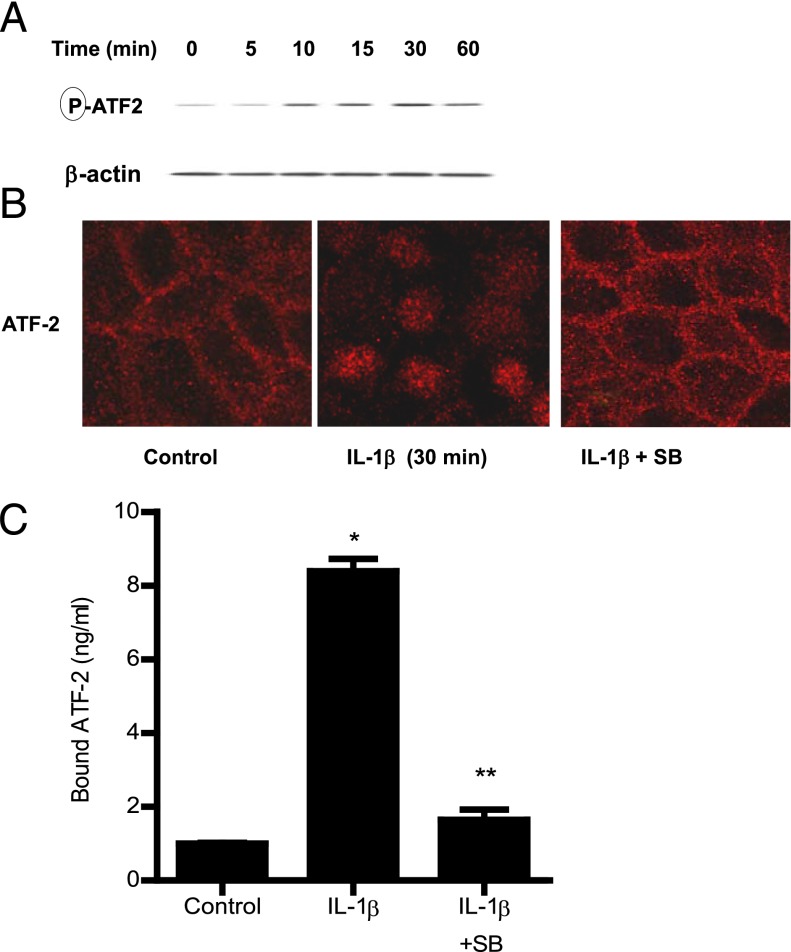
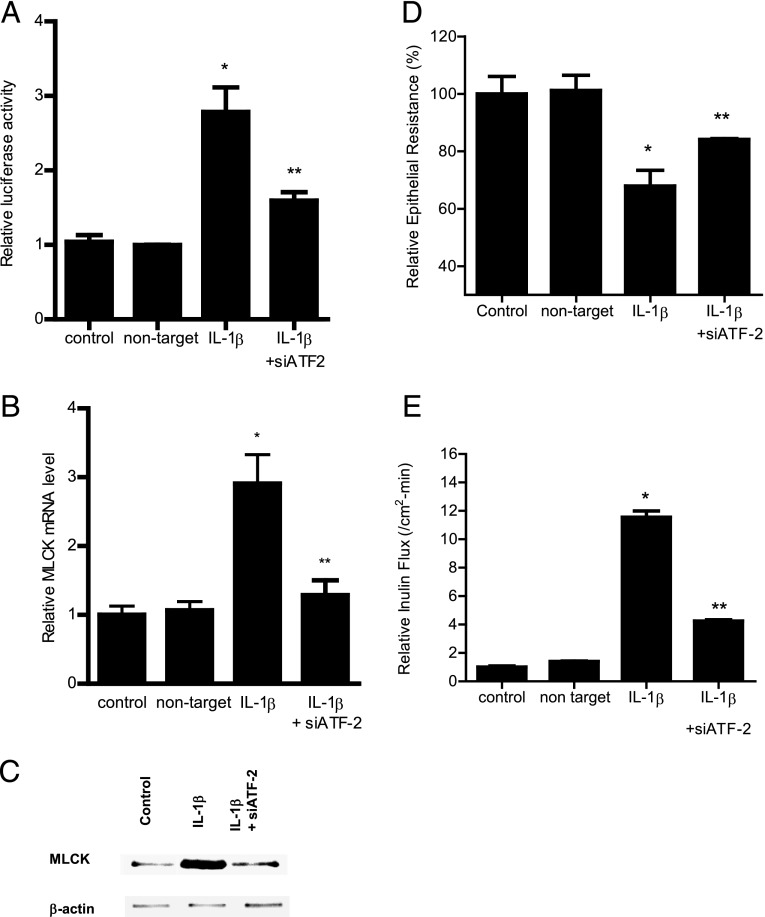
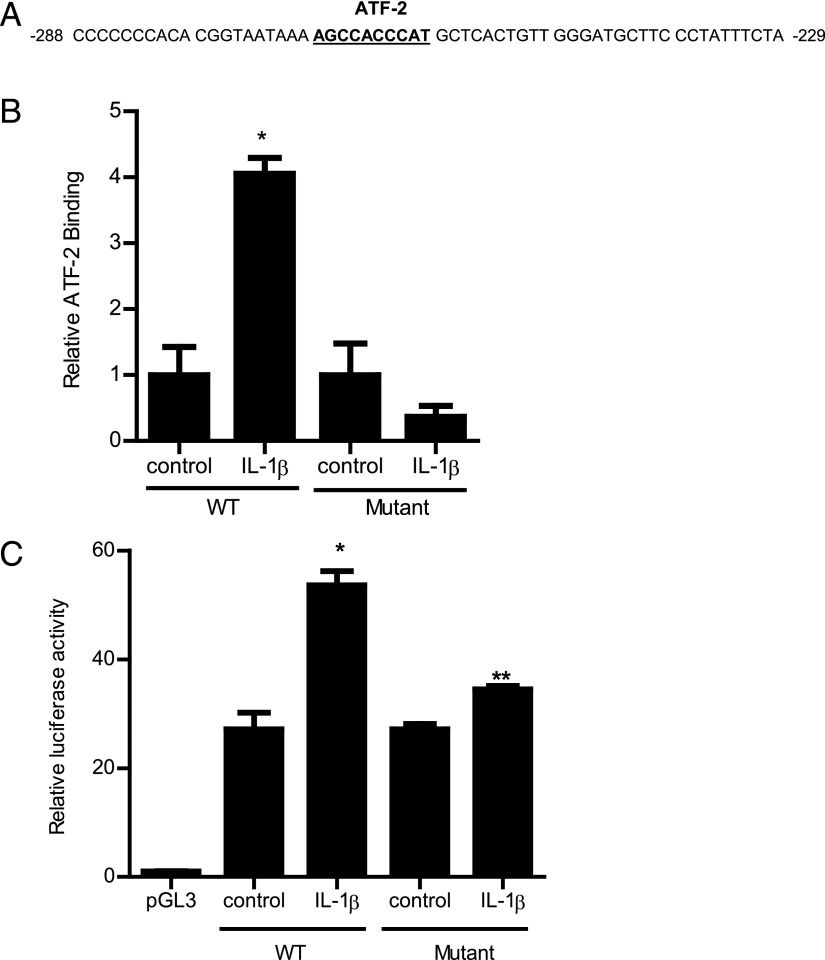
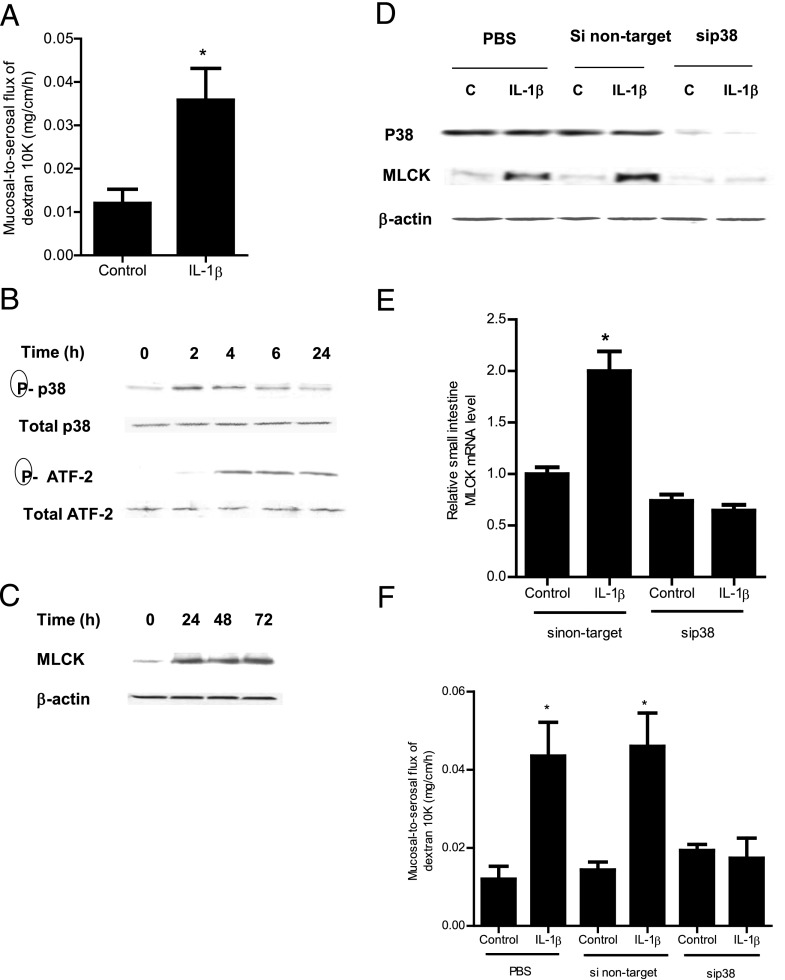
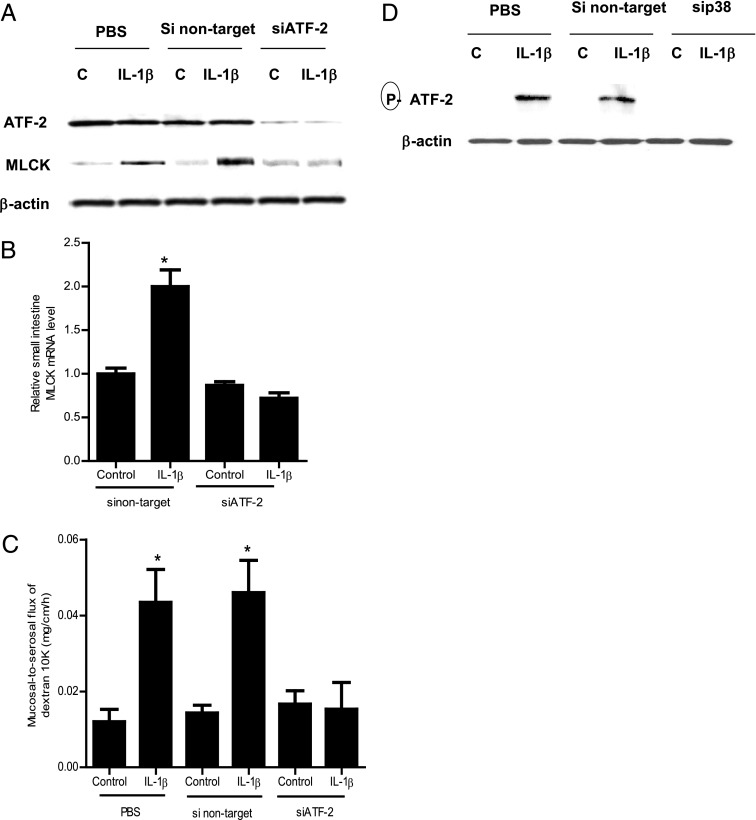
The defective intestinal epithelial tight junction (TJ) barrier has been postulated to be an important pathogenic factor contributing to intestinal inflammation. It has been shown that the proinflammatory cytokine IL-1β causes an increase in intestinal permeability; however, the signaling pathways and the molecular mechanisms involved remain unclear. The major purpose of this study was to investigate the role of the p38 kinase pathway and the molecular processes involved. In these studies, the in vitro intestinal epithelial model system (Caco-2 monolayers) was used to delineate the cellular and molecular mechanisms, and a complementary in vivo mouse model system (intestinal perfusion) was used to assess the in vivo relevance of the in vitro findings. Our data indicated that the IL-1β increase in Caco-2 TJ permeability correlated with an activation of p38 kinase. The activation of p38 kinase caused phosphorylation and activation of p38 kinase substrate, activating transcription factor (ATF)-2. The activated ATF-2 translocated to the nucleus where it attached to its binding motif on the myosin L chain kinase (MLCK) promoter region, leading to the activation of MLCK promoter activity and gene transcription. Small interfering RNA induced silencing of ATF-2, or mutation of the ATF-2 binding motif prevented the activation of MLCK promoter and MLCK mRNA transcription. Additionally, in vivo intestinal perfusion studies also indicated that the IL-1β increase in mouse intestinal permeability required p38 kinase-dependent activation of ATF-2. In conclusion, these studies show that the IL-1β-induced increase in intestinal TJ permeability in vitro and in vivo was regulated by p38 kinase activation of ATF-2 and by ATF-2 regulation of MLCK gene activity.
Figures
References
-
- Hollander D. 1999. Intestinal permeability, leaky gut, and intestinal disorders. Curr. Gastroenterol. Rep. 1: 410–416 - PubMed
-
- Turner J. R. 2009. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9: 799–809 - PubMed
-
- Ma T. Y., Anderson J. M. 2012. Tight junctions and the intestinal barrier. In Physiology of the Gastrointestinal Tract. Johnson L., ed. Elsevier Academic Press, Burlington, MA, p. 1043–1088.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
