

Molecular evolution of human adenoviruses
- PMID: 23657240
- PMCID: PMC3648800
- DOI: 10.1038/srep01812
Molecular evolution of human adenoviruses
Abstract
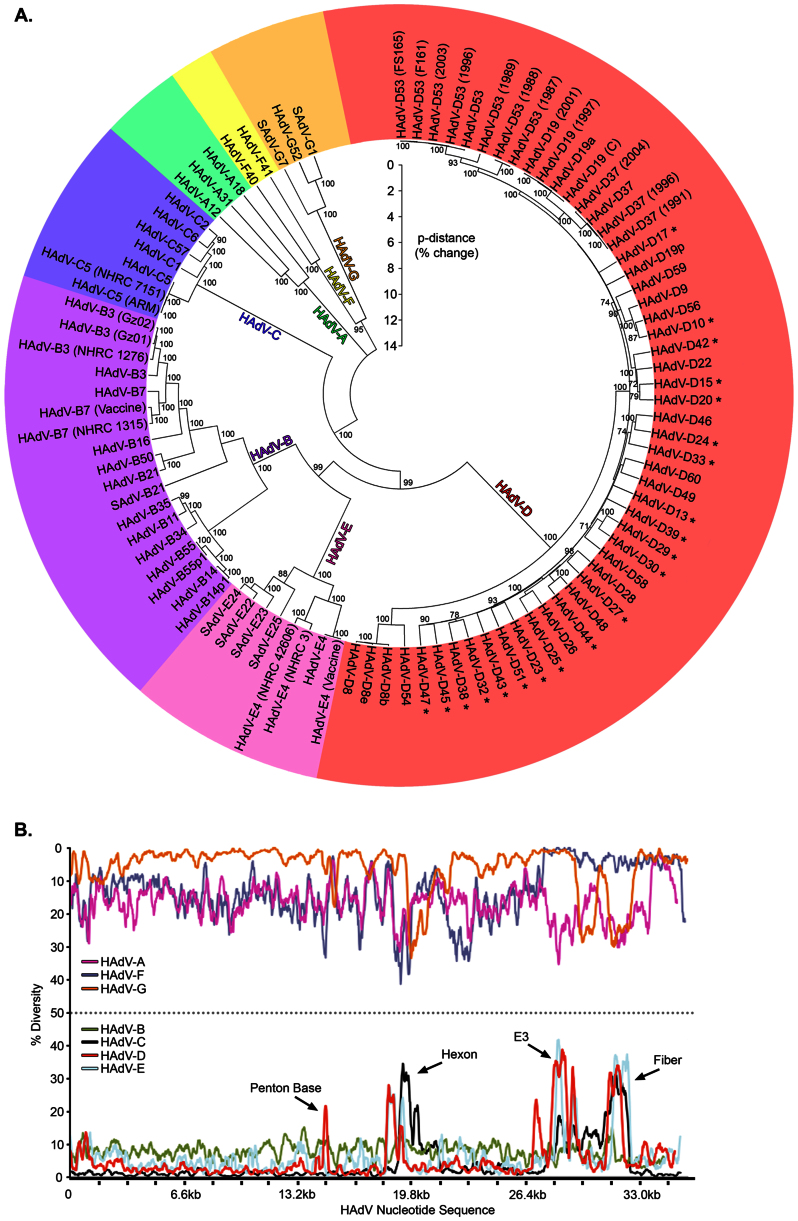
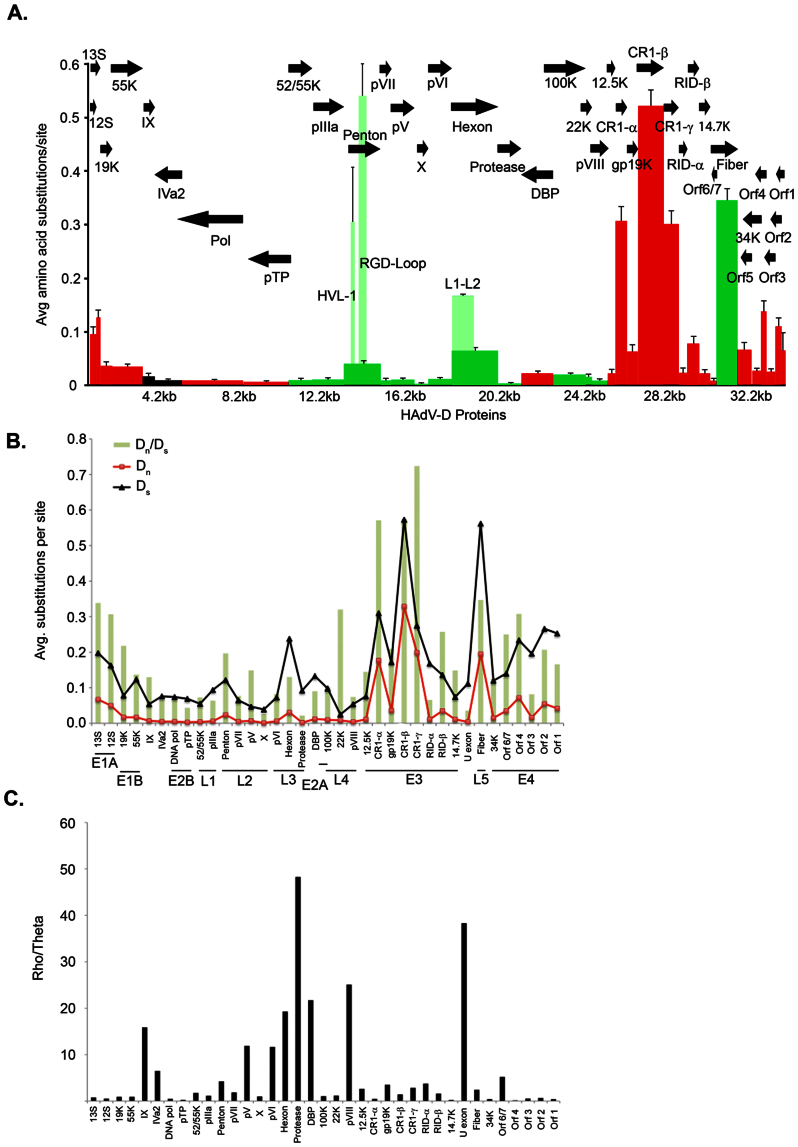
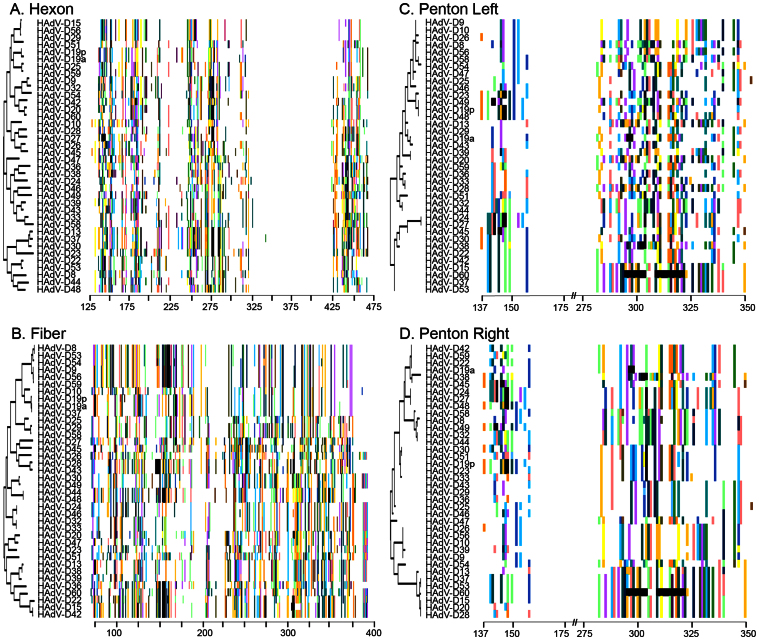
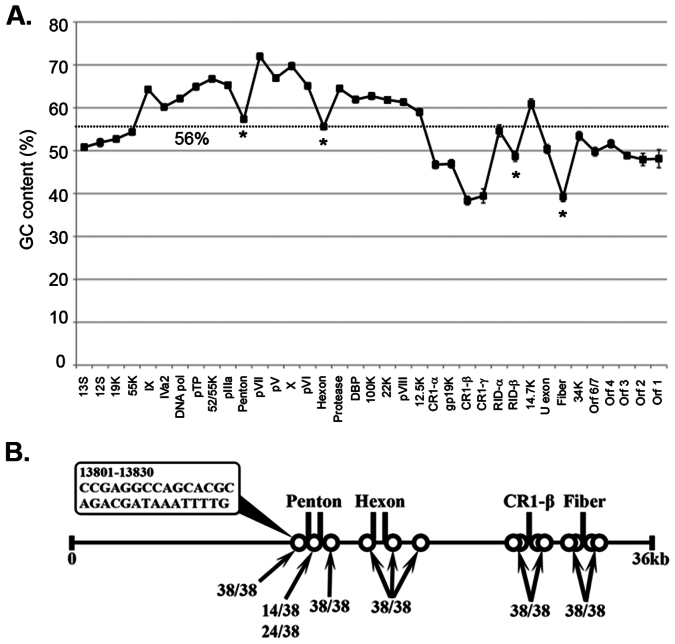
The recent emergence of highly virulent human adenoviruses (HAdVs) with new tissue tropisms underscores the need to determine their ontogeny. Here we report complete high quality genome sequences and analyses for all the previously unsequenced HAdV serotypes (n = 20) within HAdV species D. Analysis of nucleotide sequence variability for these in conjunction with another 40 HAdV prototypes, comprising all seven HAdV species, confirmed the uniquely hypervariable regions within species. The mutation rate among HAdV-Ds was low when compared to other HAdV species. Homologous recombination was identified in at least two of five examined hypervariable regions for every virus, suggesting the evolution of HAdV-Ds has been highly dependent on homologous recombination. Patterns of alternating GC and AT rich motifs correlated well with hypervariable region recombination sites across the HAdV-D genomes, suggesting foci of DNA instability lead to formulaic patterns of homologous recombination and confer agility to adenovirus evolution.
Figures
References
-
- Chow L. T., Gelinas R. E., Broker T. R. & Roberts R. J. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12, 1–8 (1977). - PubMed
-
- Whyte P. et al. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334, 124–9 (1988). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
