

Detection of mixed infection from bacterial whole genome sequence data allows assessment of its role in Clostridium difficile transmission
- PMID: 23658511
- PMCID: PMC3642043
- DOI: 10.1371/journal.pcbi.1003059
Detection of mixed infection from bacterial whole genome sequence data allows assessment of its role in Clostridium difficile transmission
Abstract
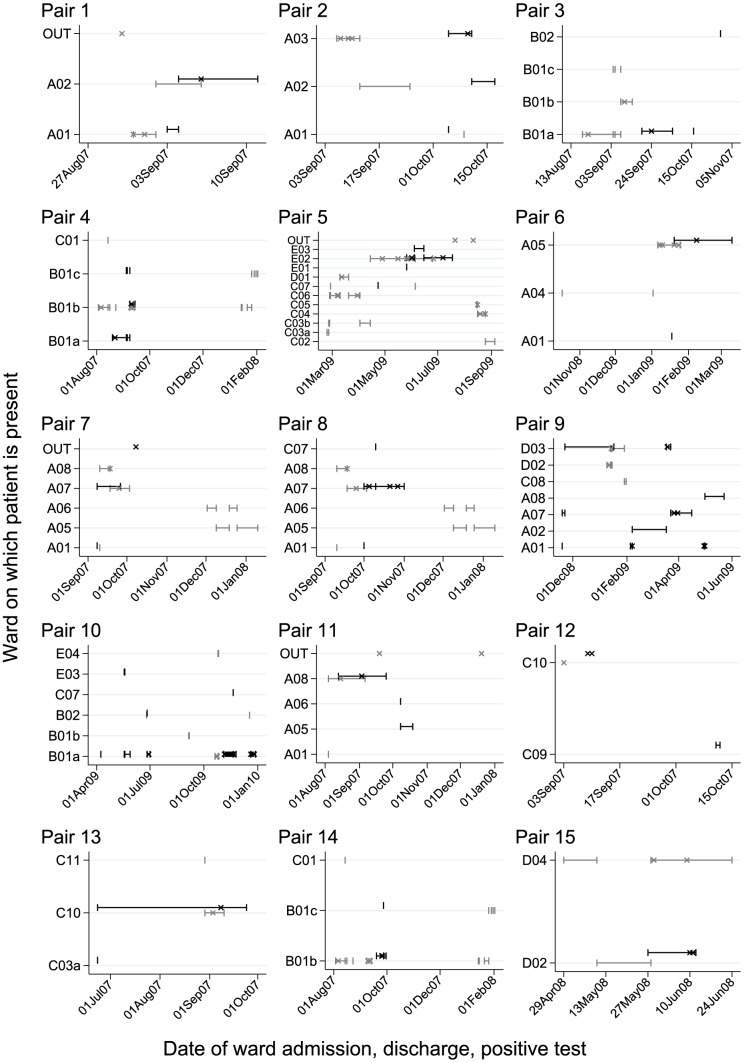
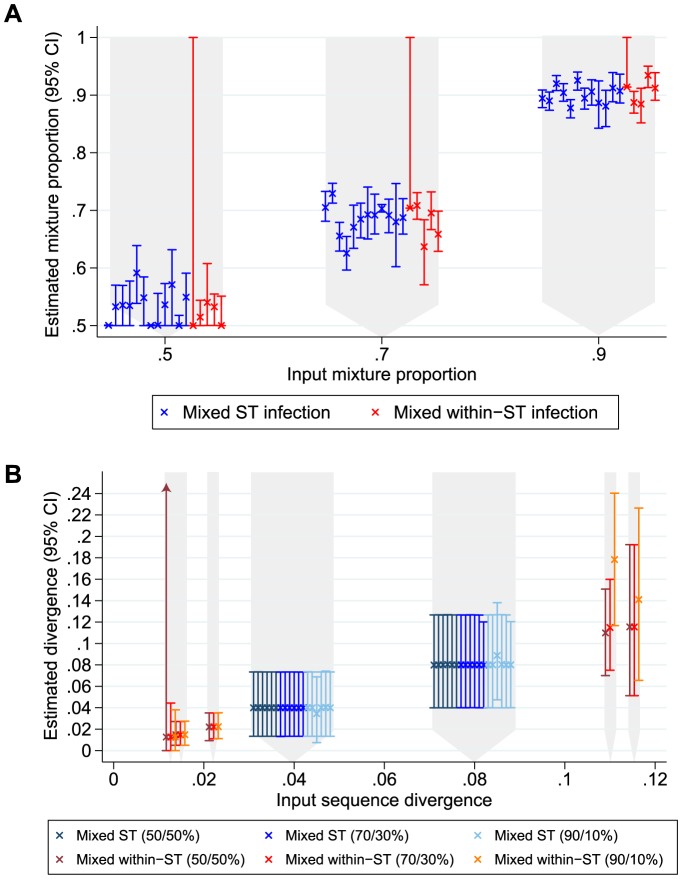
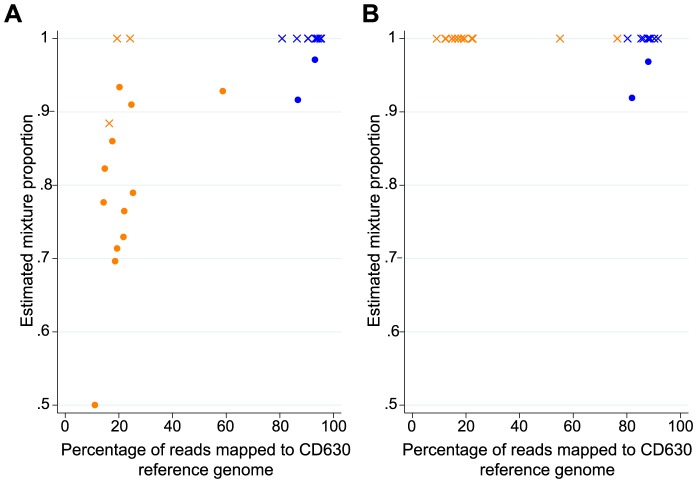
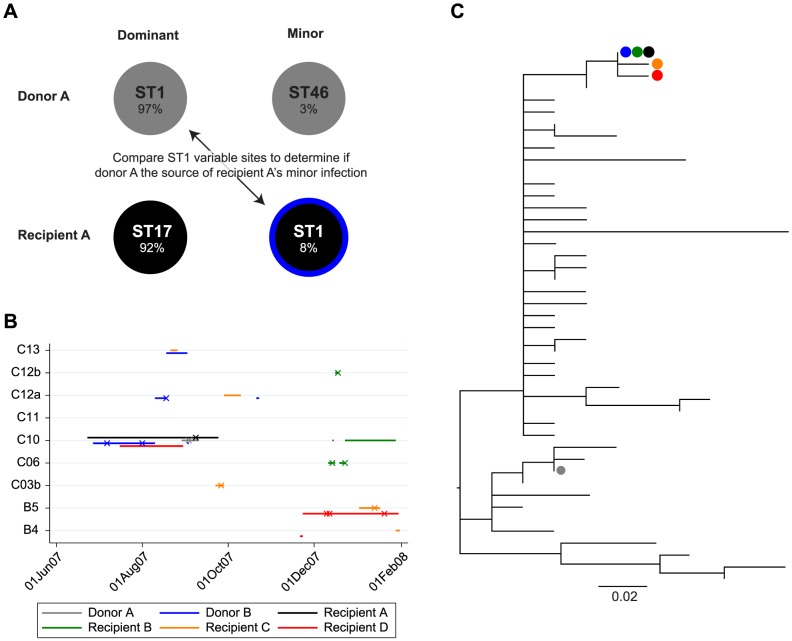
Bacterial whole genome sequencing offers the prospect of rapid and high precision investigation of infectious disease outbreaks. Close genetic relationships between microorganisms isolated from different infected cases suggest transmission is a strong possibility, whereas transmission between cases with genetically distinct bacterial isolates can be excluded. However, undetected mixed infections-infection with ≥2 unrelated strains of the same species where only one is sequenced-potentially impairs exclusion of transmission with certainty, and may therefore limit the utility of this technique. We investigated the problem by developing a computationally efficient method for detecting mixed infection without the need for resource-intensive independent sequencing of multiple bacterial colonies. Given the relatively low density of single nucleotide polymorphisms within bacterial sequence data, direct reconstruction of mixed infection haplotypes from current short-read sequence data is not consistently possible. We therefore use a two-step maximum likelihood-based approach, assuming each sample contains up to two infecting strains. We jointly estimate the proportion of the infection arising from the dominant and minor strains, and the sequence divergence between these strains. In cases where mixed infection is confirmed, the dominant and minor haplotypes are then matched to a database of previously sequenced local isolates. We demonstrate the performance of our algorithm with in silico and in vitro mixed infection experiments, and apply it to transmission of an important healthcare-associated pathogen, Clostridium difficile. Using hospital ward movement data in a previously described stochastic transmission model, 15 pairs of cases enriched for likely transmission events associated with mixed infection were selected. Our method identified four previously undetected mixed infections, and a previously undetected transmission event, but no direct transmission between the pairs of cases under investigation. These results demonstrate that mixed infections can be detected without additional sequencing effort, and this will be important in assessing the extent of cryptic transmission in our hospitals.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Genome-Based Infection Tracking Reveals Dynamics of Clostridium difficile Transmission and Disease Recurrence.Clin Infect Dis. 2016 Mar 15;62(6):746-752. doi: 10.1093/cid/civ1031. Epub 2015 Dec 18. Clin Infect Dis. 2016. PMID: 26683317 Free PMC article.
-
A global to local genomics analysis of Clostridioides difficile ST1/RT027 identifies cryptic transmission events in a northern Arizona healthcare network.Microb Genom. 2019 Jul;5(7):e000271. doi: 10.1099/mgen.0.000271. Epub 2019 May 20. Microb Genom. 2019. PMID: 31107202 Free PMC article.
-
Whole-Genome Sequencing Reveals the High Nosocomial Transmission and Antimicrobial Resistance of Clostridioides difficile in a Single Center in China, a Four-Year Retrospective Study.Microbiol Spectr. 2022 Feb 23;10(1):e0132221. doi: 10.1128/spectrum.01322-21. Epub 2022 Jan 12. Microbiol Spectr. 2022. PMID: 35019676 Free PMC article.
-
Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.Cochrane Database Syst Rev. 2022 Feb 1;2(2022):CD014217. doi: 10.1002/14651858.CD014217. Cochrane Database Syst Rev. 2022. PMID: 36321557 Free PMC article.
-
Clostridium difficile and One Health.Clin Microbiol Infect. 2020 Jul;26(7):857-863. doi: 10.1016/j.cmi.2019.10.023. Epub 2019 Nov 1. Clin Microbiol Infect. 2020. PMID: 31682985 Review.
Cited by
-
Comparative Genomics of Clostridioides difficile.Adv Exp Med Biol. 2024;1435:199-218. doi: 10.1007/978-3-031-42108-2_10. Adv Exp Med Biol. 2024. PMID: 38175477
-
Identifying Mixed Mycobacterium tuberculosis Infection and Laboratory Cross-Contamination during Mycobacterial Sequencing Programs.J Clin Microbiol. 2018 Oct 25;56(11):e00923-18. doi: 10.1128/JCM.00923-18. Print 2018 Nov. J Clin Microbiol. 2018. PMID: 30209183 Free PMC article.
-
Mycobacterium intracellulare subsp. chimaera from Cardio Surgery Heating-Cooling Units and from Clinical Samples in Israel Are Genetically Unrelated.Pathogens. 2021 Oct 27;10(11):1392. doi: 10.3390/pathogens10111392. Pathogens. 2021. PMID: 34832548 Free PMC article.
-
BHap: a novel approach for bacterial haplotype reconstruction.Bioinformatics. 2019 Nov 1;35(22):4624-4631. doi: 10.1093/bioinformatics/btz280. Bioinformatics. 2019. PMID: 31004480 Free PMC article.
-
Bacterial Genomics Reveal the Complex Epidemiology of an Emerging Pathogen in Arctic and Boreal Ungulates.Front Microbiol. 2016 Nov 7;7:1759. doi: 10.3389/fmicb.2016.01759. eCollection 2016. Front Microbiol. 2016. PMID: 27872617 Free PMC article.
References
-
- Didelot X, Bowden R, Wilson DJ, Peto TEA, Crook DW (2012) Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 13: 601–612 doi:10.1038/nrg3226. - DOI - PMC - PubMed
-
- Wilson DJ (2012) Insights from genomics into bacterial pathogen populations. PLoS Pathog 8: e1002874 doi:10.1371/journal.ppat.1002874. - DOI - PMC - PubMed
-
- Rohde H, Qin J, Cui Y, Li D, Loman NJ, et al. (2011) Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. N Engl J Med 365: 718–724 doi:10.1056/NEJMoa1107643. - DOI - PubMed
-
- Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, et al. (2011) Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N Engl J Med 365: 709–717 doi:10.1056/NEJMoa1106920. - DOI - PMC - PubMed
-
- Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, et al. (2011) Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS ONE 6: e22751 doi:10.1371/journal.pone.0022751. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
