

Sequencing platform and library preparation choices impact viral metagenomes
- PMID: 23663384
- PMCID: PMC3655917
- DOI: 10.1186/1471-2164-14-320
Sequencing platform and library preparation choices impact viral metagenomes
Abstract
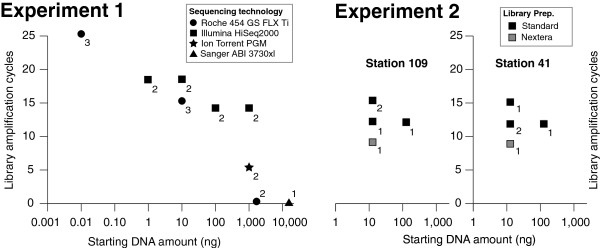
Background: Microbes drive the biogeochemistry that fuels the planet. Microbial viruses modulate their hosts directly through mortality and horizontal gene transfer, and indirectly by re-programming host metabolisms during infection. However, our ability to study these virus-host interactions is limited by methods that are low-throughput and heavily reliant upon the subset of organisms that are in culture. One way forward are culture-independent metagenomic approaches, but these novel methods are rarely rigorously tested, especially for studies of environmental viruses, air microbiomes, extreme environment microbiology and other areas with constrained sample amounts. Here we perform replicated experiments to evaluate Roche 454, Illumina HiSeq, and Ion Torrent PGM sequencing and library preparation protocols on virus metagenomes generated from as little as 10 pg of DNA.
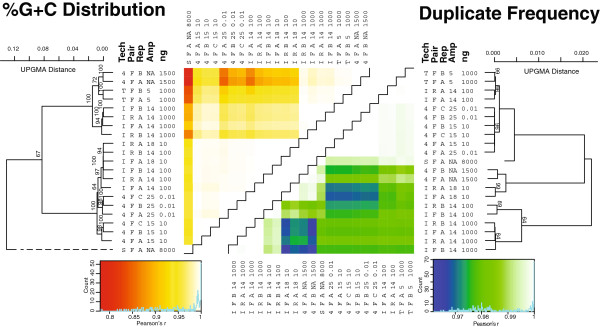
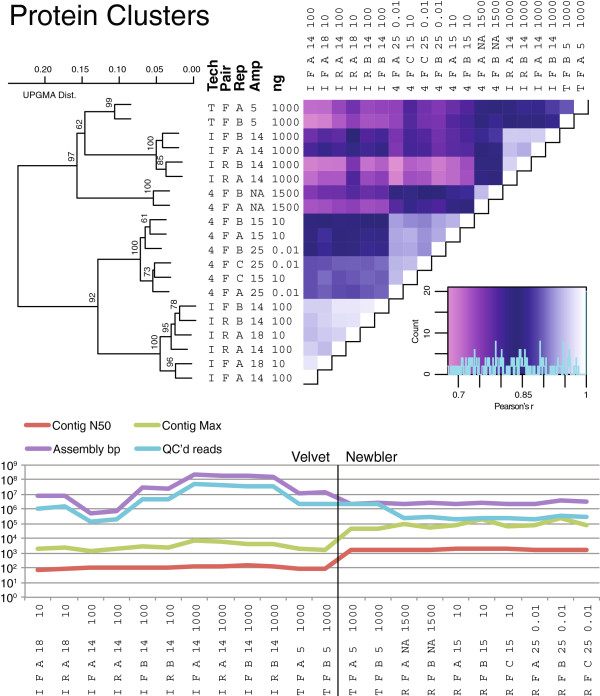
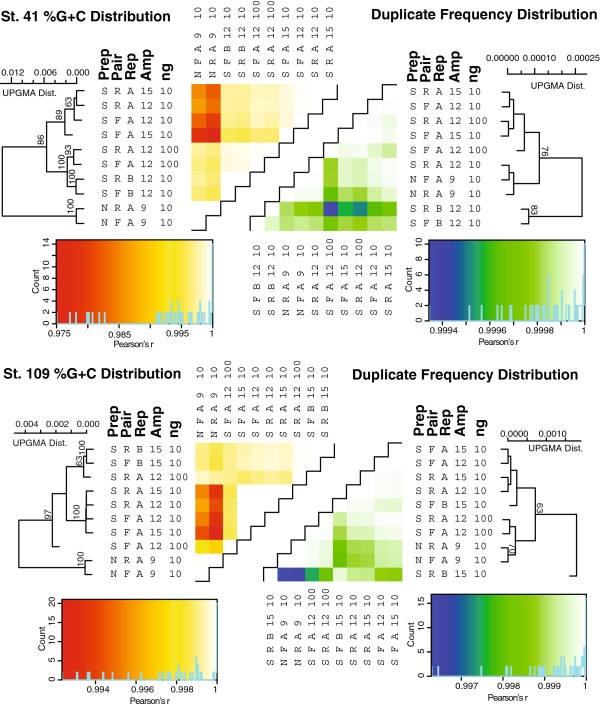
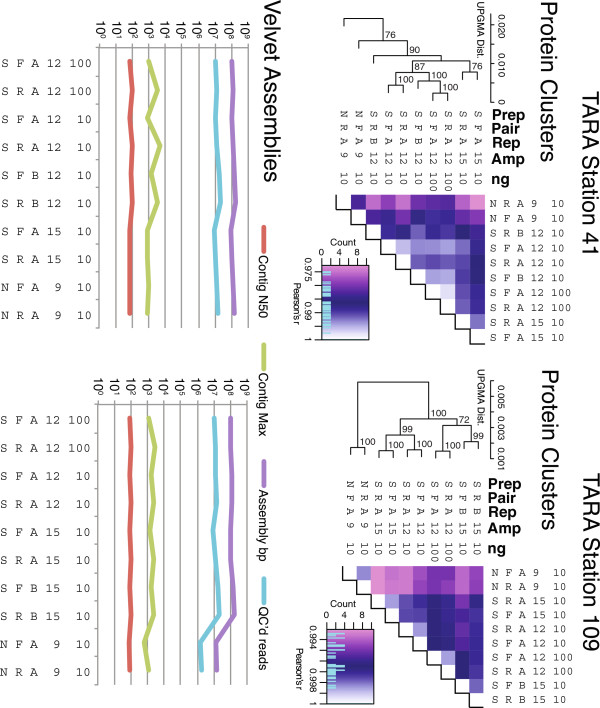
Results: Using %G+C content to compare metagenomes, we find that (i) metagenomes are highly replicable, (ii) some treatment effects are minimal, e.g., sequencing technology choice has 6-fold less impact than varying input DNA amount, and (iii) when restricted to a limited DNA concentration (<1 μg), changing the amount of amplification produces little variation. These trends were also observed when examining the metagenomes for gene function and assembly performance, although the latter more closely aligned to sequencing effort and read length than preparation steps tested. Among Illumina library preparation options, transposon-based libraries diverged from all others and adaptor ligation was a critical step for optimizing sequencing yields.
Conclusions: These data guide researchers in generating systematic, comparative datasets to understand complex ecosystems, and suggest that neither varied amplification nor sequencing platforms will deter such efforts.
Figures
References
-
- Handelsman J, Tiedje JM, Alvarez-Cohen L, Ashburner M, Cann IKO, Delong EF, Doolittle WF, Fraser-Liggett CM, Godzik A, Gordon JI. New Science of Metagenomics: Revealing the Secrets of Our Microbial Planet. Nat Res Council Report. 2007. p. 13.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
