

Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy
- PMID: 23664116
- PMCID: PMC3675237
- DOI: 10.1016/j.ajhg.2013.04.011
Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy
Abstract
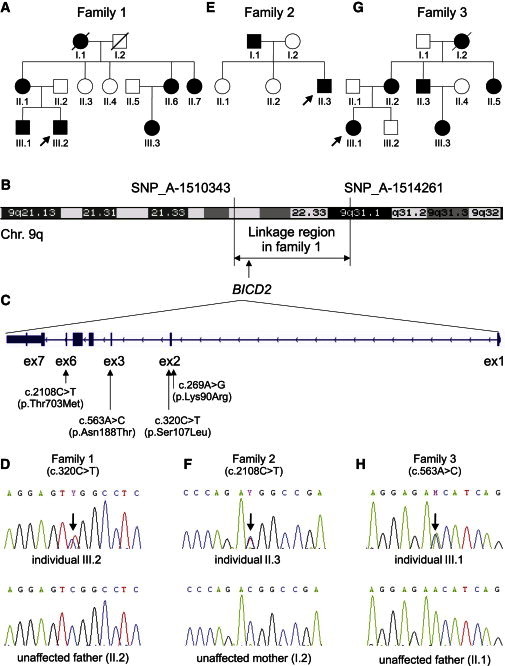
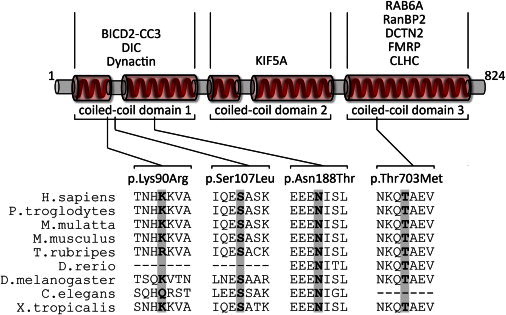
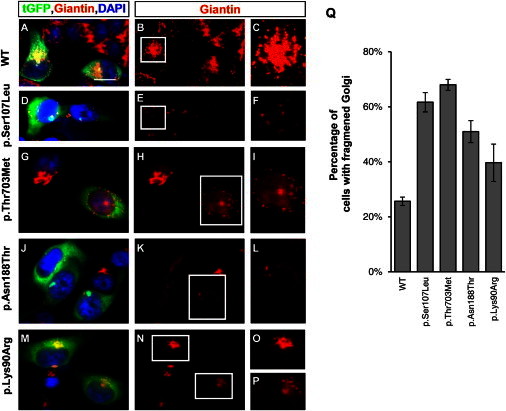
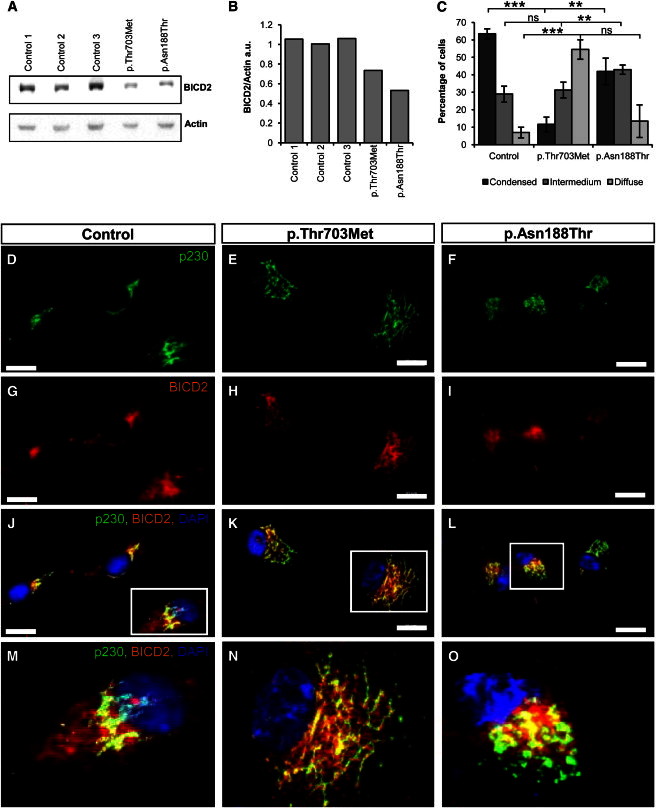
Spinal muscular atrophy (SMA) is a heterogeneous group of neuromuscular disorders caused by degeneration of lower motor neurons. Although functional loss of SMN1 is associated with autosomal-recessive childhood SMA, the genetic cause for most families affected by dominantly inherited SMA is unknown. Here, we identified pathogenic variants in bicaudal D homolog 2 (Drosophila) (BICD2) in three families afflicted with autosomal-dominant SMA. Affected individuals displayed congenital slowly progressive muscle weakness mainly of the lower limbs and congenital contractures. In a large Dutch family, linkage analysis identified a 9q22.3 locus in which exome sequencing uncovered c.320C>T (p.Ser107Leu) in BICD2. Sequencing of 23 additional families affected by dominant SMA led to the identification of pathogenic variants in one family from Canada (c.2108C>T [p.Thr703Met]) and one from the Netherlands (c.563A>C [p.Asn188Thr]). BICD2 is a golgin and motor-adaptor protein involved in Golgi dynamics and vesicular and mRNA transport. Transient transfection of HeLa cells with all three mutant BICD2 cDNAs caused massive Golgi fragmentation. This observation was even more prominent in primary fibroblasts from an individual harboring c.2108C>T (p.Thr703Met) (affecting the C-terminal coiled-coil domain) and slightly less evident in individuals with c.563A>C (p.Asn188Thr) (affecting the N-terminal coiled-coil domain). Furthermore, BICD2 levels were reduced in affected individuals and trapped within the fragmented Golgi. Previous studies have shown that Drosophila mutant BicD causes reduced larvae locomotion by impaired clathrin-mediated synaptic endocytosis in neuromuscular junctions. These data emphasize the relevance of BICD2 in synaptic-vesicle recycling and support the conclusion that BICD2 mutations cause congenital slowly progressive dominant SMA.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- De Jonghe P., Auer-Grumbach M., Irobi J., Wagner K., Plecko B., Kennerson M., Zhu D., De Vriendt E., Van Gerwen V., Nicholson G. Autosomal dominant juvenile amyotrophic lateral sclerosis and distal hereditary motor neuronopathy with pyramidal tract signs: synonyms for the same disorder? Brain. 2002;125:1320–1325. - PubMed
-
- Zickler D., Kleckner N. Meiotic chromosomes: integrating structure and function. Annu. Rev. Genet. 1999;33:603–754. - PubMed
-
- Kiernan M.C., Vucic S., Cheah B.C., Turner M.R., Eisen A., Hardiman O., Burrell J.R., Zoing M.C. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. - PubMed
-
- Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
