

Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance
- PMID: 23664119
- PMCID: PMC3675262
- DOI: 10.1016/j.ajhg.2013.04.013
Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance
Abstract
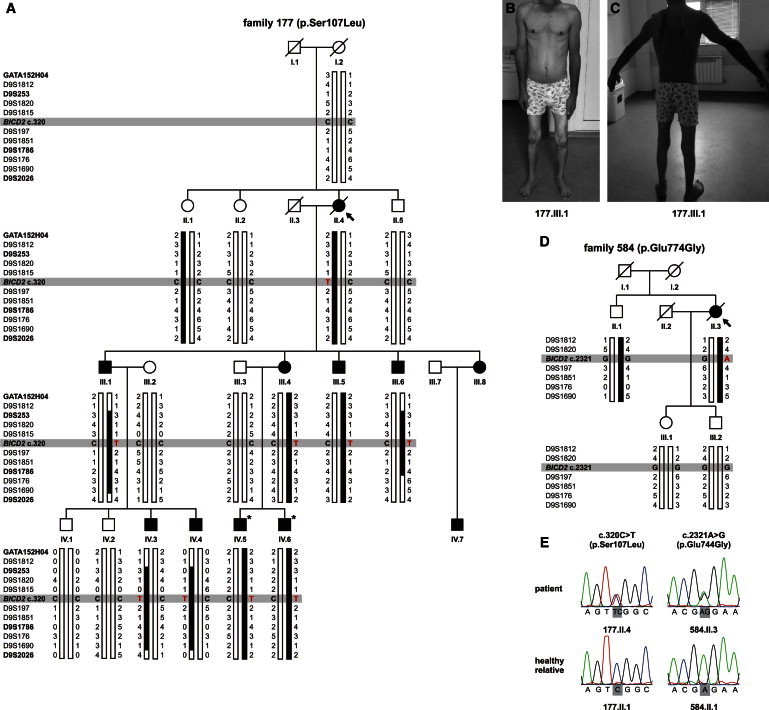
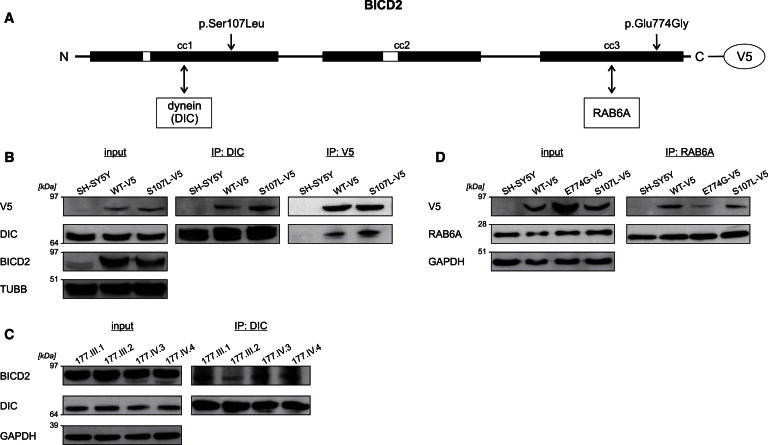
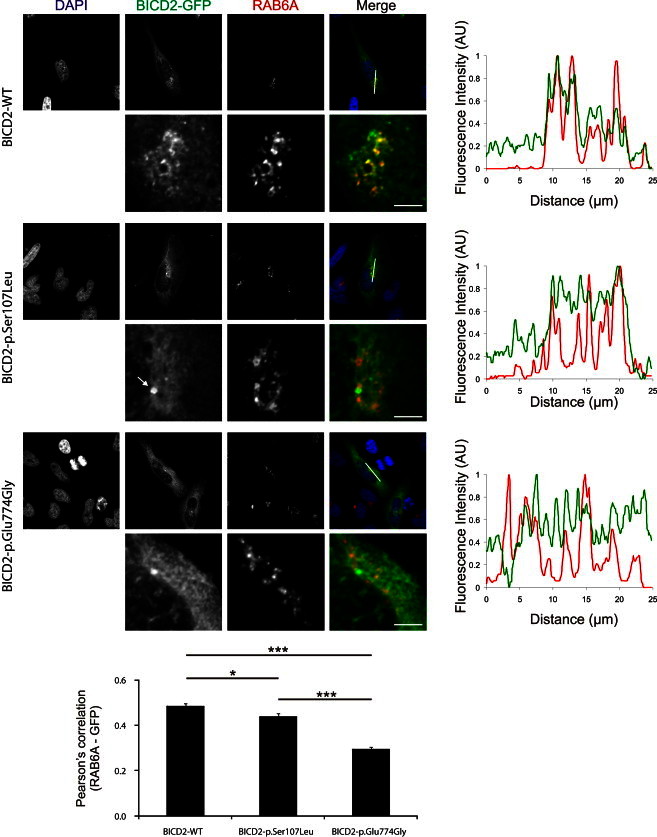
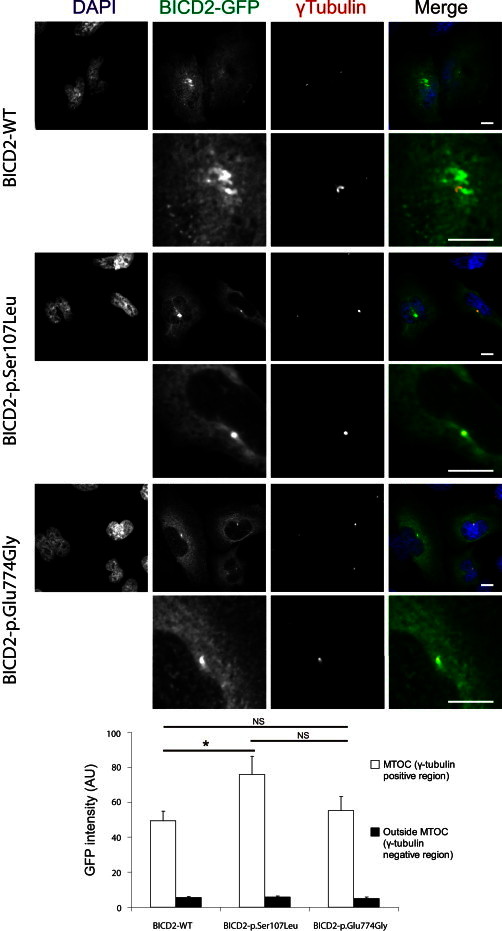
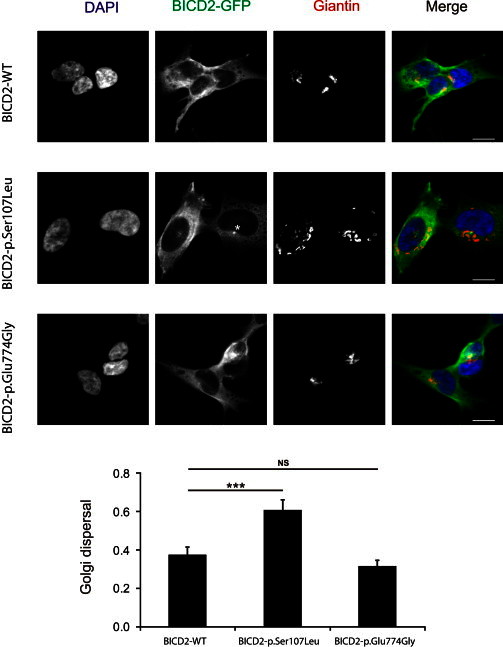
The most common form of spinal muscular atrophy (SMA) is a recessive disorder caused by deleterious SMN1 mutations in 5q13, whereas the genetic etiologies of non-5q SMA are very heterogeneous and largely remain to be elucidated. In a Bulgarian family affected by autosomal-dominant proximal SMA, we performed genome-wide linkage analysis and whole-exome sequencing and found a heterozygous de novo c.320C>T (p.Ser107Leu) mutation in bicaudal D homolog 2 (Drosophila) (BICD2). Further analysis of BICD2 in a cohort of 119 individuals with non-5q SMA identified a second de novo BICD2 mutation, c.2321A>G (p.Glu774Gly), in a simplex case. Detailed clinical and electrophysiological investigations revealed that both families are affected by a very similar disease course, characterized by early childhood onset, predominant involvement of lower extremities, and very slow disease progression. The amino acid substitutions are located in two interaction domains of BICD2, an adaptor protein linking the dynein molecular motor with its cargo. Our immunoprecipitation and localization experiments in HeLa and SH-SY5Y cells and affected individuals' lymphoblasts demonstrated that p.Ser107Leu causes increased dynein binding and thus leads to accumulation of BICD2 at the microtubule-organizing complex and Golgi fragmentation. In addition, the altered protein had a reduced colocalization with RAB6A, a regulator of vesicle trafficking between the Golgi and the endoplasmic reticulum. The interaction between p.Glu744Gly altered BICD2 and RAB6A was impaired, which also led to their reduced colocalization. Our study identifies BICD2 mutations as a cause of non-5q linked SMA and highlights the importance of dynein-mediated motility in motor neuron function in humans.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy.Am J Hum Genet. 2013 Jun 6;92(6):946-54. doi: 10.1016/j.ajhg.2013.04.011. Epub 2013 May 9. Am J Hum Genet. 2013. PMID: 23664116 Free PMC article.
-
Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2.Brain. 2015 Feb;138(Pt 2):293-310. doi: 10.1093/brain/awu356. Epub 2014 Dec 14. Brain. 2015. PMID: 25497877 Free PMC article.
-
Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia.Am J Hum Genet. 2013 Jun 6;92(6):965-73. doi: 10.1016/j.ajhg.2013.04.018. Epub 2013 May 9. Am J Hum Genet. 2013. PMID: 23664120 Free PMC article.
-
Dominant spinal muscular atrophy is caused by mutations in BICD2, an important golgin protein.Front Neurosci. 2015 Nov 5;9:401. doi: 10.3389/fnins.2015.00401. eCollection 2015. Front Neurosci. 2015. PMID: 26594138 Free PMC article. Review.
-
The Genotypic and Phenotypic Spectrum of BICD2 Variants in Spinal Muscular Atrophy.Ann Neurol. 2020 Apr;87(4):487-496. doi: 10.1002/ana.25704. Ann Neurol. 2020. PMID: 32057122 Review.
Cited by
-
Neurogenetic fetal akinesia and arthrogryposis: genetics, expanding genotype-phenotypes and functional genomics.J Med Genet. 2021 Sep;58(9):609-618. doi: 10.1136/jmedgenet-2020-106901. Epub 2020 Oct 15. J Med Genet. 2021. PMID: 33060286 Free PMC article.
-
Adapter Proteins for Opposing Motors Interact Simultaneously with Nuclear Pore Protein Nup358.Biochemistry. 2019 Dec 17;58(50):5085-5097. doi: 10.1021/acs.biochem.9b00907. Epub 2019 Dec 6. Biochemistry. 2019. PMID: 31756096 Free PMC article.
-
A Structural Model for the Core Nup358-BicD2 Interface.Biomolecules. 2023 Sep 26;13(10):1445. doi: 10.3390/biom13101445. Biomolecules. 2023. PMID: 37892127 Free PMC article.
-
Microscopic and Biochemical Hallmarks of BICD2-Associated Muscle Pathology toward the Evaluation of Novel Variants.Int J Mol Sci. 2023 Apr 6;24(7):6808. doi: 10.3390/ijms24076808. Int J Mol Sci. 2023. PMID: 37047781 Free PMC article.
-
Inherited Paediatric Motor Neuron Disorders: Beyond Spinal Muscular Atrophy.Neural Plast. 2017;2017:6509493. doi: 10.1155/2017/6509493. Epub 2017 May 28. Neural Plast. 2017. PMID: 28634552 Free PMC article. Review.
References
-
- Dubowitz V. Ramblings in the history of spinal muscular atrophy. Neuromuscul. Disord. 2009;19:69–73. - PubMed
-
- Viollet L., Barois A., Rebeiz J.G., Rifai Z., Burlet P., Zarhrate M., Vial E., Dessainte M., Estournet B., Kleinknecht B. Mapping of autosomal recessive chronic distal spinal muscular atrophy to chromosome 11q13. Ann. Neurol. 2002;51:585–592. - PubMed
-
- Takashima H., Nakagawa M., Suehara M., Saito M., Saito A., Kanzato N., Matsuzaki T., Hirata K., Terwilliger J.D., Osame M. Gene for hereditary motor and sensory neuropathy (proximal dominant form) mapped to 3q13.1. Neuromuscul. Disord. 1999;9:368–371. - PubMed
-
- Nishimura A.L., Mitne-Neto M., Silva H.C., Richieri-Costa A., Middleton S., Cascio D., Kok F., Oliveira J.R., Gillingwater T., Webb J. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004;75:822–831. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
