

Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia
- PMID: 23664120
- PMCID: PMC3675232
- DOI: 10.1016/j.ajhg.2013.04.018
Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia
Abstract
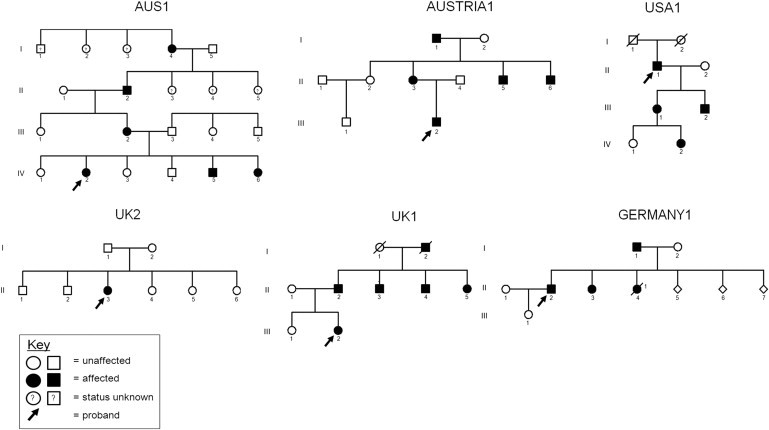
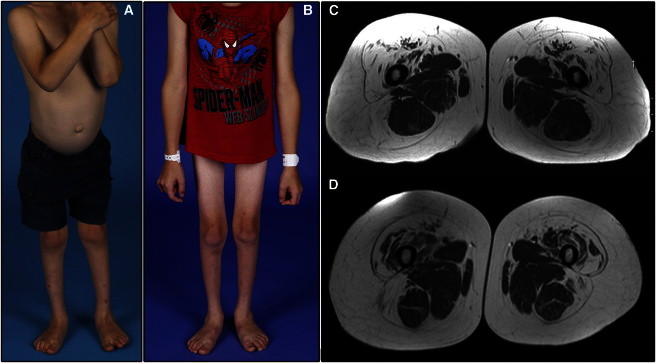
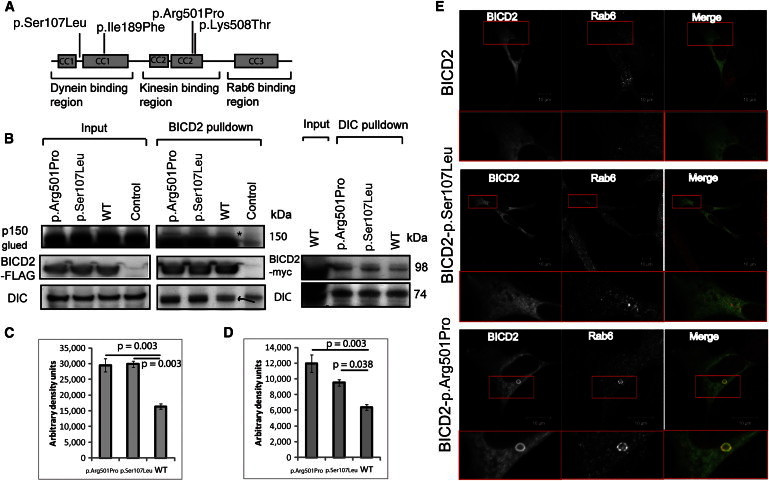
Dominant congenital spinal muscular atrophy (DCSMA) is a disorder of developing anterior horn cells and shows lower-limb predominance and clinical overlap with hereditary spastic paraplegia (HSP), a lower-limb-predominant disorder of corticospinal motor neurons. We have identified four mutations in bicaudal D homolog 2 (Drosophila) (BICD2) in six kindreds affected by DCSMA, DCSMA with upper motor neuron features, or HSP. BICD2 encodes BICD2, a key adaptor protein that interacts with the dynein-dynactin motor complex, which facilitates trafficking of cellular cargos that are critical to motor neuron development and maintenance. We demonstrate that mutations resulting in amino acid substitutions in two binding regions of BICD2 increase its binding affinity for the cytoplasmic dynein-dynactin complex, which might result in the perturbation of BICD2-dynein-dynactin-mediated trafficking, and impair neurite outgrowth. These findings provide insight into the mechanism underlying both the static and the slowly progressive clinical features and the motor neuron pathology that characterize BICD2-associated diseases, and underscore the importance of the dynein-dynactin transport pathway in the development and survival of both lower and upper motor neurons.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2.Brain. 2015 Feb;138(Pt 2):293-310. doi: 10.1093/brain/awu356. Epub 2014 Dec 14. Brain. 2015. PMID: 25497877 Free PMC article.
-
Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance.Am J Hum Genet. 2013 Jun 6;92(6):955-64. doi: 10.1016/j.ajhg.2013.04.013. Epub 2013 May 9. Am J Hum Genet. 2013. PMID: 23664119 Free PMC article.
-
Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy.Am J Hum Genet. 2013 Jun 6;92(6):946-54. doi: 10.1016/j.ajhg.2013.04.011. Epub 2013 May 9. Am J Hum Genet. 2013. PMID: 23664116 Free PMC article.
-
Dominant spinal muscular atrophy is caused by mutations in BICD2, an important golgin protein.Front Neurosci. 2015 Nov 5;9:401. doi: 10.3389/fnins.2015.00401. eCollection 2015. Front Neurosci. 2015. PMID: 26594138 Free PMC article. Review.
-
Dynein activators and adaptors at a glance.J Cell Sci. 2019 Mar 15;132(6):jcs227132. doi: 10.1242/jcs.227132. J Cell Sci. 2019. PMID: 30877148 Free PMC article. Review.
Cited by
-
De Novo and Inherited Variants in GBF1 are Associated with Axonal Neuropathy Caused by Golgi Fragmentation.Am J Hum Genet. 2020 Oct 1;107(4):763-777. doi: 10.1016/j.ajhg.2020.08.018. Epub 2020 Sep 15. Am J Hum Genet. 2020. PMID: 32937143 Free PMC article.
-
A Quantitative Model for BicD2/Cargo Interactions.Biochemistry. 2018 Nov 20;57(46):6538-6550. doi: 10.1021/acs.biochem.8b00987. Epub 2018 Nov 5. Biochemistry. 2018. PMID: 30345745 Free PMC article.
-
BICD1 mediates HIF1α nuclear translocation in mesenchymal stem cells during hypoxia adaptation.Cell Death Differ. 2019 Sep;26(9):1716-1734. doi: 10.1038/s41418-018-0241-1. Epub 2018 Nov 21. Cell Death Differ. 2019. PMID: 30464225 Free PMC article.
-
Adapter Proteins for Opposing Motors Interact Simultaneously with Nuclear Pore Protein Nup358.Biochemistry. 2019 Dec 17;58(50):5085-5097. doi: 10.1021/acs.biochem.9b00907. Epub 2019 Dec 6. Biochemistry. 2019. PMID: 31756096 Free PMC article.
-
Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies.Brain. 2014 Nov;137(Pt 11):2879-96. doi: 10.1093/brain/awu169. Epub 2014 Jun 25. Brain. 2014. PMID: 24970098 Free PMC article. Review.
References
-
- Mercuri E., Messina S., Kinali M., Cini C., Longman C., Battini R., Cioni G., Muntoni F. Congenital form of spinal muscular atrophy predominantly affecting the lower limbs: a clinical and muscle MRI study. Neuromuscul. Disord. 2004;14:125–129. - PubMed
-
- Reddel S., Ouvrier R.A., Nicholson G., Dierick I., Irobi J., Timmerman V., Ryan M.M. Autosomal dominant congenital spinal muscular atrophy—a possible developmental deficiency of motor neurones? Neuromuscul. Disord. 2008;18:530–535. - PubMed
-
- Salinas S., Proukakis C., Crosby A., Warner T.T. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. - PubMed
-
- Oates E.C., Reddel S., Rodriguez M.L., Gandolfo L.C., Bahlo M., Hawke S.H., Lamandé S.R., Clarke N.F., North K.N. Autosomal dominant congenital spinal muscular atrophy: a true form of spinal muscular atrophy caused by early loss of anterior horn cells. Brain. 2012;135:1714–1723. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54 HG003067/HG/NHGRI NIH HHS/United States
- G1001253/MRC_/Medical Research Council/United Kingdom
- G108/638/MRC_/Medical Research Council/United Kingdom
- WT091310/WT_/Wellcome Trust/United Kingdom
- P 23223/FWF_/Austrian Science Fund FWF/Austria
- MR/J004758/1/MRC_/Medical Research Council/United Kingdom
- G0802760/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- R01 NS072248/NS/NINDS NIH HHS/United States
- 1U54NS0657/NS/NINDS NIH HHS/United States
- U54 NS065712/NS/NINDS NIH HHS/United States
- MR/K000608/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
