

Transcriptional and epigenetic dynamics during specification of human embryonic stem cells
- PMID: 23664763
- PMCID: PMC3709577
- DOI: 10.1016/j.cell.2013.04.037
Transcriptional and epigenetic dynamics during specification of human embryonic stem cells
Abstract
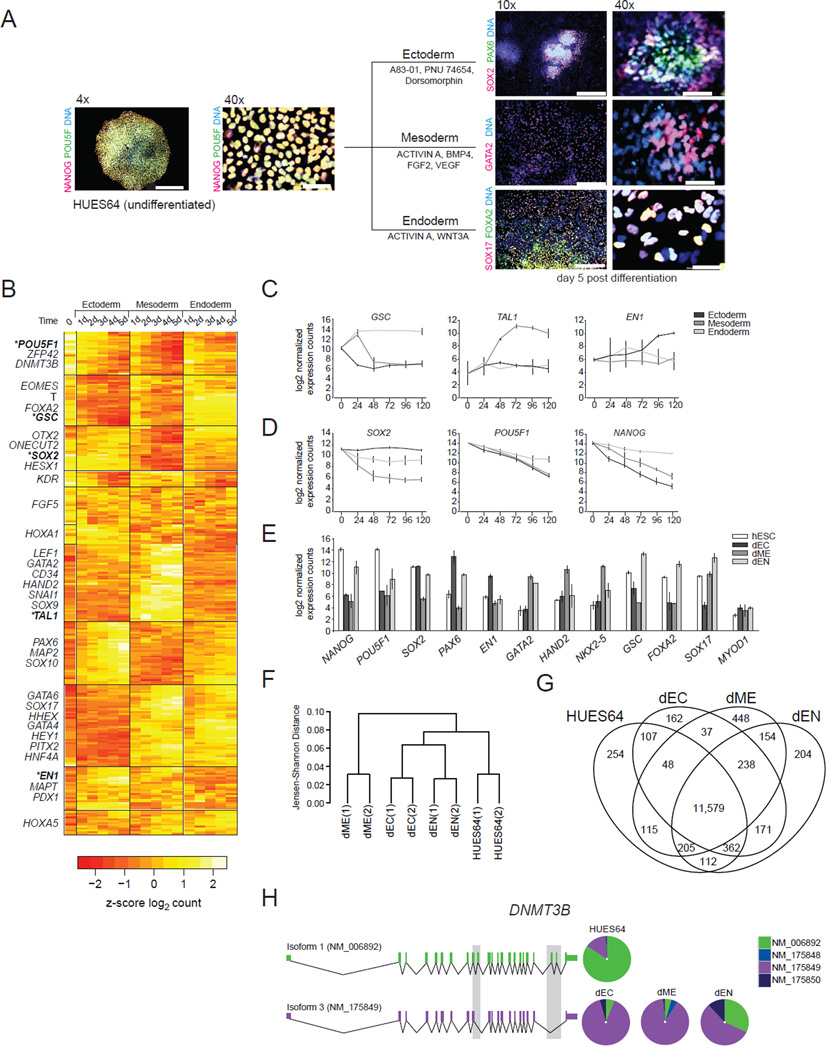
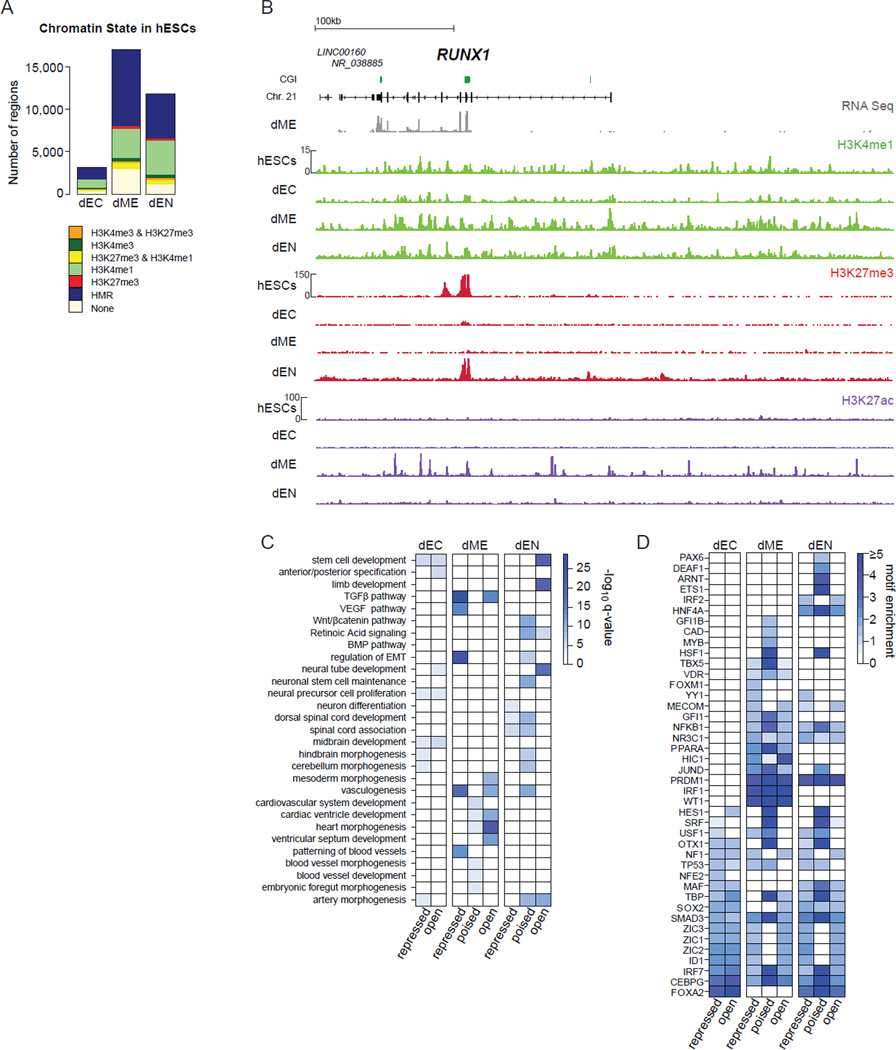
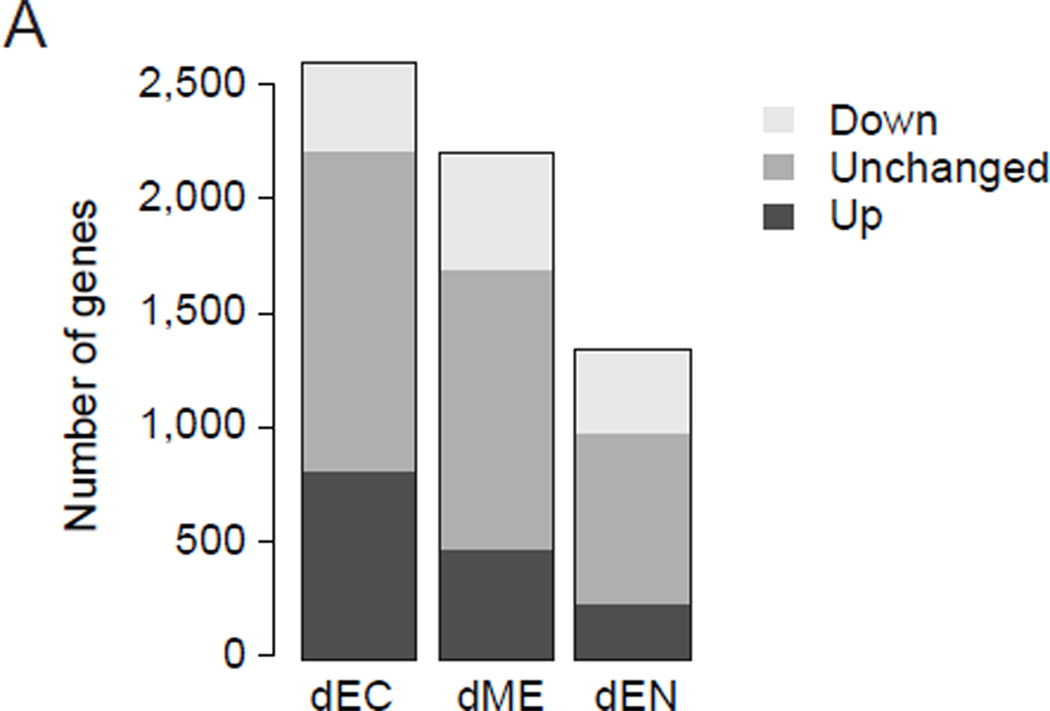
Differentiation of human embryonic stem cells (hESCs) provides a unique opportunity to study the regulatory mechanisms that facilitate cellular transitions in a human context. To that end, we performed comprehensive transcriptional and epigenetic profiling of populations derived through directed differentiation of hESCs representing each of the three embryonic germ layers. Integration of whole-genome bisulfite sequencing, chromatin immunoprecipitation sequencing, and RNA sequencing reveals unique events associated with specification toward each lineage. Lineage-specific dynamic alterations in DNA methylation and H3K4me1 are evident at putative distal regulatory elements that are frequently bound by pluripotency factors in the undifferentiated hESCs. In addition, we identified germ-layer-specific H3K27me3 enrichment at sites exhibiting high DNA methylation in the undifferentiated state. A better understanding of these initial specification events will facilitate identification of deficiencies in current approaches, leading to more faithful differentiation strategies as well as providing insights into the rewiring of human regulatory programs during cellular transitions.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures




References
-
- Ahlgren U, Pfaff SL, Jessell TM, Edlund T, Edlund H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature. 1997;385:257–260. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
